(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
Many of the comorbidities of the metabolic syndrome are now known to be due to inflammatory changes [1–4]. There is said to be a low-grade chronic systemic inflammation, and serum levels of various inflammatory cytokines, chemokines, and inflammatory markers including TNF-α; interleukins IL-1β, IL-8, and IL-10; monocyte chemoattractant protein-1 (MCP-1); macrophage inflammatory protein-1α (MIP-1α); growth-regulating oncogene-α (GRO-α); inducible protein-10 (IP-10); and C-reactive protein (CRP) are increased. Adipocytes are active secretors of various inflammatory and chemotactic cytokines, and a substantial portion, if not all, of the raised serum levels are contributed by the adipose tissue [5–7]. Obesity, systemic inflammation, and insulin resistance are linked in such a way that it is difficult to decide what comes first. Perhaps the three are in an autocatalytic loop.
Many of the comorbidities of the metabolic syndrome are now known to be due to inflammatory changes [1–4]. There is said to be a low-grade chronic systemic inflammation, and serum levels of various inflammatory cytokines, chemokines, and inflammatory markers including TNF-α; interleukins IL-1β, IL-8, and IL-10; monocyte chemoattractant protein-1 (MCP-1); macrophage inflammatory protein-1α (MIP-1α); growth-regulating oncogene-α (GRO-α); inducible protein-10 (IP-10); and C-reactive protein (CRP) are increased. Adipocytes are active secretors of various inflammatory and chemotactic cytokines, and a substantial portion, if not all, of the raised serum levels are contributed by the adipose tissue [5–7]. Obesity, systemic inflammation, and insulin resistance are linked in such a way that it is difficult to decide what comes first. Perhaps the three are in an autocatalytic loop.
At the level of ultimate cause, there is no satisfactory explanation as yet for why adipose tissues secrete chemokines. If this property has been consistently shown, it must have evolved under some kind of selective pressure. Chemokine secretion by adipocytes must be adaptive under at least certain set of conditions, although it is pathological in the context of metabolic syndrome. There have been attempts to explain ultimate causes behind the obesity-induced hypercytokinemia under the thriftiness paradigm. It has been interpreted as raised level of innate immunity [8], and its evolution is said to be a response to increased chances of infection under starvation conditions in which the thrifty phenotype evolved [9]. If starvation and infection challenges co-occurred during hunter-gatherer life, thrifty genotype and infection-resistant genotype may have coevolved. This is in support to the notion that infections accompany famines, and most of the deaths during famines are due to infections than to starvation [10, 11]. However, an inherent weakness of this explanation is that in obesity or insulin resistance, there is no evidence of increased resistance to infections. The raised levels of inflammatory cytokines have not been demonstrated to confer increased overall resistance to infections. On the contrary many specific infections, including infections of the skin and urinary tract, are more common in diabetic patients [12]. Other infections occur with increased severity and are associated with an increased risk of complications in patients with obesity and diabetes [13].
The behavioral origin hypothesis attempts to explain the inflammatory changes in a different way. In a soldier life the risk of getting wounds and injuries is substantially greater as compared to a diplomat life. Therefore the immune cells should move peripheral, i.e., towards subcutaneous tissue. The phagocytic cells of the body, namely, neutrophils and macrophages, are produced centrally in the bone marrow. They circulate in blood, macrophages in blood being in the form of monocytes. Any injury causes the extravasation, i.e., movement of cells across vessel walls and migration of these cells from local blood vessels towards the site of injury. This is the normal dynamics of these cells. In the natural ancestral environment, minor skin injuries would be common, and stimulated by such injuries, these cells will continually migrate towards the skin. There is hardly any backflow in this dynamics. But the normal dynamics would keep a steady-state density of cells in blood vessels and a certain density in subcutaneous tissue as well. Whenever there is a shift to a diplomat state, epidermal injuries are less probable and the normal dynamics would be altered. There will be reduced migration from blood vessels to skin. As a result the density of macrophages in subcutaneous tissue would be less than normal, and their density in flowing blood and presumably attached to vessel walls would increase. In other words the immune system could be retracting from the periphery and as a result would be centrally hyperactive. This results into delayed wound healing on the one hand and increased inflammatory tendency of vascular tissue and some other central organs on the other.
Can we test the hypothesis? First of all, does a negative association or a trade-off between cutaneous and central immunity exist? If yes, does it change in obesity and metabolic syndrome? If yes, what are the molecular mechanisms involved? There is some evidence for trade-off or shift of balance among the innate immune mechanisms in obesity and insulin resistance. There is a negative association between atopic allergy and obesity [14]. Cutaneous delayed-type hypersensitivity is reduced in both type 1 and type 2 diabetes [15, 16], suggesting that at least some of the components of the immune system are partially retracted from the skin. The peripheral density of macrophages [17] and mast cells [18] is reported to be reduced in diabetes. Along with macrophages, mast cells also have important functions in wound healing [19–22]. On the other hand macrophages appear to accumulate in large numbers in adipose tissue [23–26] and so do mast cells [27]. Macrophage accumulation in blood vessels is an essential and very early phase in macrovascular complications in diabetes [28]. In diabetic nephropathy, macrophage accumulation is an important contributor and arresting macrophage accumulation can prevent diabetic nephropathy. There is no overall increase in macrophages throughout the body. Macrophage accumulation is tissue specific, and certain tissues simultaneously experience dearth of macrophages [17]. It is therefore evidently a problem of altered macrophage distribution than overall rise in macrophage activity. The redistribution is due to altered migration patterns of innate immune cells. The rate of macrophage and neutrophil migration is shown to be altered in diabetes [29–32]. Therefore my conjecture of altered phagocyte dynamics in diabetes has adequate support, although the complete picture of the dynamics is yet to emerge. From available data a number of mechanisms can be reconstructed by which the dynamics might shift.
1.Testosterone: One of the major players in the immune redistribution is likely to be testosterone. Traditionally testosterone is said to be immunosuppressive. Braude et al. [33] suggested that testosterone may be an immune redistributor rather than an immunosuppressor. According to their hypothesis, testosterone brings about movement of the immunological garrison to the periphery since testosterone is associated with aggression and therefore predicts injuries. Although there have been hardly any attempts to seriously test the Braude et al. [33] hypothesis, there is some circumstantial evidence in its favor. On the one hand serum testosterone levels are negatively associated with serum inflammatory markers and other components of the metabolic syndrome [34, 35]. Testosterone is shown to be protective against inflammatory complications of the metabolic syndrome [35]. On the other hand, although testosterone is generally considered immunosuppressive, it is shown to enhance inflammation (but not wound healing) of cutaneous wounds [36–38]. Thus the action of testosterone on inflammation appears to be in opposite directions in the context of skin and that of central organs. Testosterone is proinflammatory for cutaneous wounds and anti-inflammatory at the vascular and systemic level. Although testosterone does not help in wound healing, it stimulates secretion of EGF and other growth factors that are needed for wound healing. This is what we expect to be an adaptive response to aggression-provoked injury proneness.
2.Peroxisome proliferator activator receptors (PPARs): The PPARs appear to be another major class of players in a shift of balance. PPARs arrest inflammation on the one hand [39] and enhance wound healing and wound closure on the other [40, 41]. Epidermal injuries activate the expression of PPARs and especially that of PPAR-β [41]. Absence of injuries on adopting a diplomat way of life may shift the immune balance against healing and towards systemic inflammation by downregulating PPAR expression.
3.Sensory nerves and neuropeptides: Sensory nerves and neuropeptides also play a significant role in modulating inflammation and wound healing [42–45]. In diabetes there is a progressive degeneration of peripheral nerves [46], shifting the immune balance further away from the skin.
4.Adipokines: Perhaps the most important mechanisms of altering the dynamics of phagocytes in the body are through adipokines. Movement of immune cells is under a gradient of chemokines [47–50] secreted at the site of injury by resident tissue cells stimulated by injury. The chemokines are locally retained on extracellular matrix and cell surface heparan sulfate proteoglycans, establishing a stable concentration gradient around the stimulus. Chemokine signaling activates leukocyte integrins leading to extravasation. Cells, after leaving the vessels, migrate to the site of injury guided by the chemokine gradient. The magnitude of the gradient is determined by the difference between the local level and the baseline blood level.
This is where adipokines play a major role. If the basal levels of chemokines are high, the gradient will be weak, and migration will be downregulated. Adipocyte secretion of chemokines increases the basal level and thereby arrests the flow of inflammatory cells towards periphery. Fat accumulation is a sign of a lifestyle and behavioral change, and therefore, it is the right kind of tissue to bring about the redistribution of the immune system. Diabetes is known to reduce inflammatory response to cutaneous injuries [41], and this may be due to the raised basal levels of chemokines.
The action of chemoattractants and other signaling molecules largely depends upon their environment and their relative rather than absolute concentrations. Increased expression of a signaling molecule need not always mean increased action. The local topology of its relative concentrations can be of utmost importance. Experiments have shown that the effects of local application of TNF-α are substantially different from the effects of its systemic administration. In a mice study local application of 50–500 ng of TNF-α in collagen improved wound healing, but similar quantity in phosphate-buffered saline equivalent quantities intraperitoneally did not [51]. Presumably TNF-α in collagen was able to form a stable gradient, whereas that in saline dispersed systemically too rapidly. Systemic TNF-α was unable to produce any topological structure and therefore could not help the wound healing process at all.
We will now examine using diffusion kinetics the effects of raised basal chemokine levels on the gradient formation and the chemotactic migration of cells. If we assume simple diffusion of a chemoattractant, gradient formation can be computed using Fick’s law of diffusion [43]:
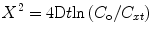 where X = distance from the origin of the chemoattractant, D = diffusion constant, t = time of diffusion, C o = concentration at the origin, and C xt = concentration at a distance X at time t.
where X = distance from the origin of the chemoattractant, D = diffusion constant, t = time of diffusion, C o = concentration at the origin, and C xt = concentration at a distance X at time t.
It can be seen that the concentration gradient thus formed is nonlinear and highly concave towards the lower end (Fig. 8.1). Let us assume that there is a threshold level of concentration difference that is necessary for the cells to recognize a gradient. A sphere (or hemisphere) of a radius equal to the distance between the source and the threshold forms the “catchment volume” of cell recruitment. The distance between the origin and threshold concentration will be the radius of this hemisphere. Therefore the catchment volume will rise in cubic proportion of X. A larger catchment volume will result in greater infiltration of cells.
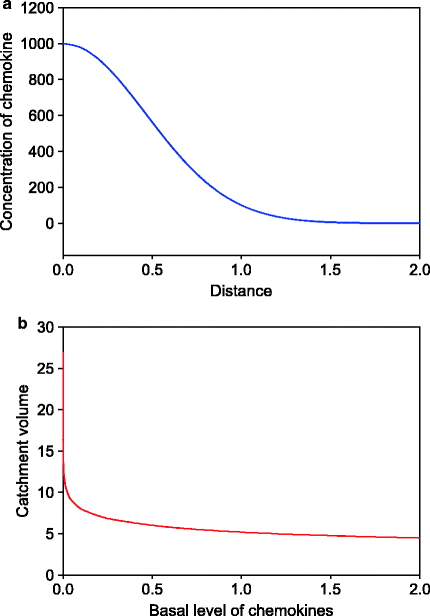
Fig. 8.1
Gradient of a chemoattractant from a site of injury and its effect on recruitment of inflammatory cells. (a) The expected diffusion gradient of a chemoattractant based on Fick’s law of diffusion. The gradient will stop at the basal plasma level of the chemoattractant. Therefore the radius from which neutrophils and/or macrophages can be recruited is decided by the basal plasma level of chemoattractant. (b) The curve shows how the catchment volume for recruiting cells changes with basal chemokine levels. At small basal levels, a very small increase reduces the catchment substantially. At large basal levels it becomes relatively insensitive to changes. The curves illustrate the principle that a small increase in the basal plasma levels of a chemoattractant would result into substantial reduction in cell recruitment at the site of injury
The threshold difference must depend upon the basal levels of the chemoattractant. If the basal levels rise, the threshold for chemotaxis should also increase. The concave nature of the concentration gradient results into a nonlinear reduction in the catchment volume such that when the threshold is small, even a little increment in it can cause a large decrement in the catchment. But when the threshold is high, a large change is required to cause the same decrement in the catchment (Fig. 8.1).
Data on normal and obesity-induced levels of TNF-α, IL-6, and MCP-1 show that there can be a two- to tenfold rise in their basal levels in obesity [52]. For example, Bastard et al. [52] found that the mean levels of TNF-α in obese patients were about double the mean for lean controls. Those for IL-6 were about tenfold. Data on their concentrations at the point of origin (C o of the above model) are difficult to obtain, but there are indications that the difference could be of many orders of magnitude as compared to the healthy basal levels [51]. Assuming a 1,000-fold difference between the source and threshold concentration, let us take the healthy basal level as 1 arbitrary unit and the source concentration as 1,000 units. Let us further assume that the threshold difference is directly proportional to the basal level with a proportionality constant of unity. With these assumptions, if the threshold levels are raised to 2 or 10 units, i.e., 0.2–1% of local levels, the model computes that there would be about 15 and 45% reduction in cell infiltration respectively. This calculation demonstrates that the reduction in infiltration is disproportionately larger. The values that we have assumed here need not match reality. As long as there is a large difference between local and systemic levels of chemokines, our conclusion will be valid. It means that a small rise in basal levels of chemokines can bring about substantial reduction in cell recruitment. Thus a small cost of raising the blood levels of chemokines can save a much greater investment in peripheral immunity. This is considerable immunological parsimony.
Interestingly, in diabetes, weakening of the gradient is brought about from the other end too. As the basal levels of chemokines are increased by adipocyte chemokine production from the other end, i.e., the site of injury, there is reduction and prolongation of chemokine production by local macrophages [17, 53–56]. Macrophage density in subcutaneous tissue is substantially reduced in diabetes [17]. This would be an autocatalytic response since a decreased density would decrease local chemokine production affecting further recruitment of macrophages. As the density of tissue macrophages goes down on the one hand, on the other, blood monocyte counts evidently go up in diabetes leading to macrovascular pathology [57–60].
A sedentary life devoid of physical activity and physical aggression creates a condition in which peripheral injuries are less likely. Obesity is prevalent under similar lifestyle. Therefore an immune reversal from the periphery under nonaggressive sedentary lifestyle mediated by adipose tissue could have evolved. The basal levels of chemokines cannot be expected to be stable. They will be subject to stochastic spatiotemporal fluctuations. The fluctuations are bound to increase in amplitude with rising basal levels. A stochastic peak exceeding the threshold can result into a nonspecific inflammatory trigger. This is a chance event that can occur anywhere in the body at any time. Therefore raised basal levels can result in stochastic inflammatory responses that can turn pathological. The modern urban lifestyle is much more sedentary and much less injury prone as compared to the conditions in which we evolved. It is possible therefore that this lifestyle acts as a supernormal stimulus for the naturally adaptive immune redistribution. An exaggerated immune redistribution can lead to vascular inflammation and its downstream pathological consequences.
There are three possible lines of work by which the hypothesis of adipokine-mediated redistribution of innate immune cells can be tested. First, it should be possible to test experimentally in animal models whether raising basal blood levels of chemokines by infusion decreases infiltration of immune cells to an experimental wound. This can be the most direct test of the hypothesis. As a long-term consequence of the altered basal levels, the distribution of immune cells in the body, mainly macrophages, is likely to change in obesity and related disorders. By appropriate labeling of macrophages followed by a whole-body scan, one could test whether the distribution of macrophages and other immune cells is different in healthy versus obese, insulin-resistant, or diabetic individuals. Also, on an epidemiological scale, it can be tested whether continued long-term exposure to minor cutaneous injuries is protective against obesity-induced inflammatory complications.
5.Signals from bone and muscle: It is well established by now that adipose tissue is an active endocrine organ and has effects on the immune system along with other systems. A less well-known but rapidly upcoming concept views bone and muscle too as endocrine organs. Osteocalcin, a molecule involved in strengthening bones, has multiple beneficial effects including insulin-sensitizing, β cell proliferative, and adiponectin-secreting actions [61–66]. Muscles also give out certain anti-inflammatory endocrine signals in the form of myokines [67–69]. This happens with every contraction of the muscle which means that the anti-inflammatory signals are directly proportional to muscle exercise. The effect of exercise on insulin resistance was earlier thought to be brought about through fat reduction. It is known now that exercise gives anti-inflammatory and insulin-sensitizing signals directly and independent of its effects on body fat. Similar to adipokines, the signal molecules released by muscle can be called “myokines.” The concept of myokines is new, and not much research inputs have gone in. But we can expect them to play a significant role in the dynamics of innate immune cells.
In addition to myokines there is a possibility of muscle-generated nervous signals as well. Muscles are rich in sensory nerve supply, and a sensory impulse is generated with every contraction of muscle. What happens to so much information being generated form muscle continuously? As argued earlier, it is possible that this information is used in the judgment of one’s own muscle strength and activity and accordingly modifying behavioral and physiological strategies. Strong muscle signifies readiness for physical aggression and would program the immune system in anticipation of injuries. Thus exercise should stimulate the dynamics of macrophages and increase their density peripherally. This would have a systemic anti-inflammatory effect, and this is what is signaled by the myokines as well as neuronal signals given by every contraction of muscle. This is why exercise has immediate effects on the dynamics and distribution of innate immune cells of the body. It has been shown that with exercise, macrophage density and function in peripheral tissues are increased [70, 71] and that in adipose tissues [72] and lungs are decreased. This is in direct support of the macrophage redistribution hypothesis. Interestingly exercise-induced redistribution leads to increased susceptibility to respiratory infections [71, 73]. This gives plausible answers to why exercise has been reported to have contradictory effects on macrophage function [70, 74].
6.Cholesterol: Cholesterol is a marker of aggression deficiency. Since the need for cutaneous innate immunity is reduced in nonaggressive lifestyle, it would not be a surprise if cholesterol arrested movement of innate immune cells towards the periphery. Cholesterol, and LDL in particular, arrests the extravasation of macrophages at the endothelial level, and this may be viewed as a normal adaptive role of blood cholesterol, which may turn pathological under supernormal aggression loss.
We have thus seen that on adopting diplomat lifestyle, the distribution of innate immune cells of the body alters substantially. The changed distribution has a major role in the pathophysiology of diabetes and related disorders. There are three main mechanisms through which the immune redistribution contributes to the pathophysiology of diabetes and related disorders.
1.
Atherosclerosis and CVD: The classical paradigm assumes LDL cholesterol to be solely responsible for the atherosclerotic changes in the arteries. What is not sufficiently appreciated is the role of macrophages in initiating these changes. Our hypothesis is that the reduced flow of monocyte–macrophages from blood to periphery leaves a larger number of macrophages within the blood vessels. Some of the excess macrophages accumulate somewhere in the vessel walls and trigger a mild localized inflammation-like response which is the starting point of the fatty streaks and progressive pathophysiological processes leading to macrovascular pathology. The role of macrophages in initial stages of fatty streaks is clearly demonstrated [28, 75]. The novel suggestion coming out of our theory is only that the macrophages that normally should have migrated to peripheral tissues but fail to do so owing to lack of injury stimulation and raised basal chemokine levels are the ones that initiate the process. Therefore aggression and epidermal injury proneness can certainly be preventive and may even reverse the lesions in the blood vessels to some extent.
There is a paradox regarding inflammation and atherosclerosis and cardiovascular disease that the orthodox theory cannot explain but the new one may. Atherosclerosis and cardiovascular disease are said to be inflammatory disorders, and there is a wide-scale agreement on this [3, 75]. Glucocorticoids are known for their strong anti-inflammatory action and are successful therapeutic agents against a number of other inflammatory conditions [76–79]. However they do not appear to be useful in atherosclerosis and CVD. In fact high cortisol levels are positively associated with atherosclerosis and CVD and are considered risk factors [80–84]. Testosterone is also considered to be an immunosuppressive and anti-inflammatory agent, and it has protective effects against atherosclerosis and CVD [85–92]. If a generalized inflammatory tendency was responsible for initiating atherogenic changes, then any anti-inflammatory agent should have been sufficient to prevent it. But cortisol and testosterone appear to have opposite effects in this context, although both are said to be anti-inflammatory. This means that it is the immune redistribution and not generalized inflammation that is important. The altered distribution of macrophages can have proinflammatory effects in certain tissues and anti-inflammatory in others. This also implies that both testosterone and cortisol are not generalized anti-inflammatory agents as is usually believed. Both are differential immune redistributors, and as a result, they have differential pro- and anti-inflammatory effects in different tissues. The proaggression hormone testosterone drives the macrophages peripheral and reduces their vascular accumulation, whereas the anti-aggression cortisol is expected to have the opposite effect.
2.
Oxidative stress: Oxidative stress is believed to be the single largest contributor to the pathological processes leading to diabetic complications. The most common source of reactive oxygen species (ROS) in healthy life is the phagocytes of the body. Unlike the popular perception, ROS production is not primarily a pathological process. It has important functions in normal metabolism and cell functions. In phagocytes of the body ROS are important weapons to kill bacteria that enter through an open injury [93]. When epidermal injuries become uncommon and the distribution of phagocytes changes, the distribution of ROS in the body is also bound to change. Since phagocytes remain within central circulation in greater numbers, there will be greater circulating concentrations of ROS. This is further enhanced by the chronic activation of these cells by inflammatory signals given by adipokines and partly by local inflammatory foci themselves. Therefore the increased oxidative stress in diabetes could be a direct consequence of the altered dynamics of the innate immune cells driven by a non-injury-prone diplomat lifestyle.
3.
Dysfunctional wound healing and angiogenesis: Apart from their phagocytic role, macrophages have other important roles in the process of wound healing. Decreased macrophage density in the peripheral tissues is a likely cause of delayed wound healing [17]. A process of great importance during wound healing is the process of blood vessel formation called angiogenesis [94]. Macrophages have an important role in the initiation of angiogenesis [95]. Macrophages activated by inflammation or hypoxia can initiate neovascularization [96–98]. This suggests an interesting possibility that the adipocyte-derived chemokines that attract macrophages have a selfish role. Growth of the adipose tissue needs angiogenesis [99, 100]. The macrophage population attracted and activated by adipokines may have a crucial role in the growth of adipose tissue by facilitating angiogenesis. Without angiogenesis the growing adipose tissue will not get sufficient supply of glucose and fatty acids to accumulate fat. A possible inference from this is that the primary role of adipokines may be in facilitating growth of adipose tissue itself through facilitated angiogenesis [101], and the systemic inflammatory changes is only a side effect seen under supernormal adipogenesis. However, the amount of adipokines produced and the number of macrophages attracted are substantially higher than what might be needed for adipose tissue angiogenesis. Adipokines could be simultaneously playing both the roles, that of facilitating a selfish local angiogenesis and that of achieving immunological parsimony for the whole body too.
Angiogenesis dysfunction is a major contributor to the pathophysiology of diabetes and, without the above logic, is full of apparent contradictions. One of the important factors causing impaired wound healing in diabetes is decreased angiogenesis, and any attempts to facilitate angiogenesis hasten the healing process [102–105]. It is interesting to note that most proaggression and sex signals have a proangiogenic action too, including testosterone, estradiol, and DHEA [106, 107]. This indicates that anticipation of injuries and preparation towards healing are inherently built in the process of aggression itself. Contrasting with this it is excessive and unwanted angiogenesis that leads to diabetic nephropathy, and attempts to arrest angiogenesis are helpful in arresting nephropathy [108–110]. The role of vascular endothelial growth factor (VEGF) in impaired wound healing and diabetic nephropathy is diametrically opposite. This paradox can be resolved by considering the macrophage redistribution. In tissues where the macrophage density is low, there is subnormal angiogenesis, and tissues where they accumulate, there is supernormal angiogenesis. Macrophage accumulation is shown to be one of the earliest events in diabetic nephropathy [111–113]. Thus, interestingly, both subnormal and supernormal angiogeneses contribute to diabetic complications simultaneously. The orthodox paradigm has not attempted to resolve this paradox, whereas the aggression deficiency hypothesis has logical and empirically supported explanation for both the extremes of angiogenesis simultaneously occurring in the body.
The central role of chronic low-grade systemic inflammation in T2D is quite well known. My interpretation of the causes and the process of inflammation is new. This is supported by a number of studies that show anti-inflammatory action of proaggression signals and vice versa. Although there are not many studies, brain serotonin, the mediator of aggression suppression, has proinflammatory effect in arthritis and depletion of serotonin ameliorates arthritis [114, 115] whereas dopamine appears to have anti-inflammatory actions [116, 117]. Melatonin also has proaggression action and is simultaneously anti-inflammatory and pain modulating [118, 119]. The anti-inflammatory properties of testosterone are already discussed, but even the female sex-related hormones estrogens are anti-inflammatory [120–123]. DHEA also has anti-inflammatory properties [124–126]. Effect of DHEA on angiogenesis is contradictory and dose dependent [127]. Although at higher concentrations DHEA inhibited in vitro endothelial cell proliferation, at concentration found in blood, it enhanced vascular endothelial proliferation [128]. Exogenous application of DHEA accelerated wound healing [129]. Apart from aggression signals, our hypothesis expects that frequent minor injuries or stimulation of the skin that generates minor but frequent local inflammations will have long-term systemic anti-inflammatory effects. Currently, we do not have data to test this, but there are some suggestive pieces of evidence. Many arthropod bites and stings have systemic anti-inflammatory effects; bee and scorpion venoms have been studied in some details, and a number of mechanisms for which are speculated [130–136]. Venom therapy has also been tried for inflammatory disorders. Acupuncture also has demonstrated anti-inflammatory effects, although the mechanisms may be debated [137, 138]. From our hypothesis’ point of view, any injury-like skin stimulation would have systemic anti-inflammatory effects which are brought about by immune redistribution. When there is increased flow of phagocytes towards the skin, systemic inflammation presumably gets suppressed. This is certainly testable but has not been tested as yet.
We need to compare the two alternative interpretations of inflammatory tendency in T2D, that by the classical versus the new hypothesis. The classical view expects thrift to be accompanied by increased immunity. There appears to be no evidence for “thrift” being associated with increased immunity. The raised inflammatory markers in T2D do not signify higher levels of immunity. In fact the risk of certain types of infections appears to be significantly more. Interestingly not all types of infections seem to be more common in diabetics [13, 139]. The question why only certain systems become more infection prone and not others has not been addressed by the classical stream of thought.
The new interpretation does not talk about increased or decreased immunity. It rather postulates an altered distribution of immunity. In diabetes the innate immune mechanisms are partially withdrawn from the periphery. As a result there would be decreased immunity towards wound infections and other peripheral infections. The organs that undergo degenerative changes also may become more susceptible to infections in diabetes. However, there is no reason why upper respiratory or gastrointestinal infections should be more common in diabetics. In fact, although diplomats might be less prone to epidermal injuries, their more social and indoor life might make them more prone to respiratory infections. Therefore our hypothesis would expect that in anticipation, the respiratory immunity may be more enhanced in diplomats. In chimpanzees dominant individuals were found to have lower levels of circulating immunoglobulins and to be more susceptible to respiratory infections [140]. This is certainly testable in humans too, but testing this from published data is difficult. Positive associations between disorders are frequently reported, but there is a research bias as well as publication bias against negative associations. Also comparing frequencies of infections to reflect susceptibility to infections must be accompanied by careful control for factors affecting pathogen exposure. For example, individuals with sedentary lifestyle are more prone to diabetes, and we expect them to be less susceptible to respiratory infections, but their lifestyle may expose them more towards crowded indoors which are transmission-promoting environments for respiratory infections. Therefore it would be dangerous to compare from available literature. A study can be carefully designed to minimize confounding factors and then ask whether diabetics are more or less prone to upper respiratory infections. There are some data showing that they are more prone to lung infections [141–143], but lungs do undergo microangiopathy in diabetes [144–148] and that can be an added complication. No diabetes-related pathology in the upper respiratory tract is known, so it would be a good comparison. I can make a testable prediction here that although diabetics may get some specific infections more frequently, the frequency of upper respiratory infections would be at par or even lower than age-matched controls after correcting for other confounding factors. One large sample study has shown that in Asians, death due to respiratory infections decreases monotonically with obesity parameters. Unlike CVD and many other causes of death, it does not show a U-shaped curve with obesity [149]. Many other studies also show that upper respiratory infections in diabetics are not different or are less common than nondiabetic controls [141, 142]. However, inferring from published data being inadequate, one should wait for a specific study addressing this question. In addition to respiratory infections weight gain appears to have a negative association with gastrointestinal disease [150]. Whether obesity and insulin resistance offers better protection from gastrointestinal infections also needs to be investigated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


