Fig. 1
The primary cytokines involved in atherosclerosis. In red: atherogenic cytokines. In green: atheroprotective cytokines. In blue: exact role still under investigation
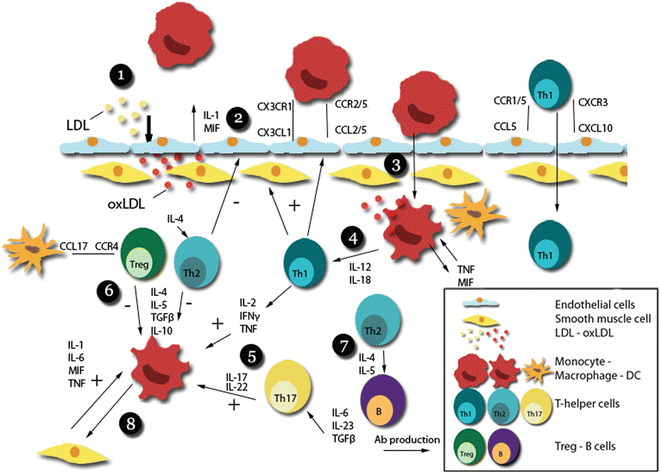
Fig. 2
Cytokines in the immune response of atherosclerosis. (1) LDL enters the vessel wall, and transforms into oxLDL by the action of enzymes and reactive oxygen species. (2) oxLDL induces immune cell chemotaxis and adhesion by endothelial cells. (3) Adhesion and diapedesis of monocytes and subsequent transformation into oxLDL-phagocyting macrophages producing autocrine cytokines such as TNF and MIF. (4) Aggravation of the initial immune response via macrophage-T-cell interactions propagating a Th1-based immune response via IL-12 and IL-18. (5) Th1-related cytokines such as IFNγ, but also IL-17 produced by Th17 cells, exacerbate the immune response by further stimulating macrophages. (6) Th2 cells and Tregs produce anti-inflammatory cytokines such as IL-10, dampening the immune response. (7) Th2 cells, formed under the influence of IL-4, induce B-cell proliferation and antibody production via IL-4 and 5. (8) Further exacerbation of the immune response occurs via macrophage-SMC interactions
In this review, we aim to give a structural overview of the cytokines involved in atherosclerosis (Table 1), with special focus on the interleukin family and mouse studies.
Table 1
Overview of cytokines in mouse atherosclerosis models
Cytokine | Receptor | Models | Diet | Outcome | Reference |
|---|---|---|---|---|---|
IL-1α | IL-1r | IL-1α−/− | AD | – Atherosclerosis↓ | [2] |
IL-1α−/−, induction | CC | – NS neointima | [182] | ||
IL-1α−/−, apoE−/− | CC | – Atherosclerosis↓ | [3] | ||
IL-1α+/−, apoE−/− | CC | – Atherosclerosis↓ | [3] | ||
IL-1α/β−/−, ldlr−/− | AD | – Atherosclerosis↓ | [11] | ||
apoE−/−, IL-1α vaccine(IL-1α-C-Qβ) | AD | – Atherosclerosis↓ | [4] | ||
IL-1α−/−, ldlr−/− | AD | – Atherosclerosis↓ | [5] | ||
IL-1β | IL-1r(1/2) | IL-1β−/− | AD | – NS | [2] |
IL-1β−/−, induction | CC | – Neointima↓ | [182] | ||
IL-1β−/−, apoE−/− | CC | – Atherosclerosis↓ | |||
IL-1β−/−, ldlr−/− | AD | – NS | [5] | ||
apoE−/−, anti-IL-1β (XMA052 MG1K) | AD | – Atherosclerosis↓ | [7] | ||
IL-1r1 | IL-1r1−/−, induction | CC | – Neointima↓ | [182] | |
IL-1r1−/−, apoE−/− | AD/CC | – Atherosclerosis↓ | |||
AD(WD) | – NS | [184] | |||
CC | – NS | [184] | |||
IL-1r1−/−, apoE+/− | AD | – Atherosclerosis↓ | [186] | ||
CC | – NS | [186] | |||
IL-1r1−/−, apoE+/−, induction | AD/CC | – Atherosclerosis↓ | [186] | ||
IL-1Ra | IL-1r | IL-1Ra−/− | AD | – NS | [187] |
IL-1Ra−/−, induction | CC | – Neointima↑ | [188] | ||
IL-1Ra treatment, induction | CC | – Neointima↓ | [182] | ||
IL-1Ra−/−, apoE−/− | CC | – Atherosclerosis↑ | [189] | ||
IL-1Ra+/−, apoE−/− | CC | – Atherosclerosis↑ | [189] | ||
IL-1RaTg, ldlr−/− | AD | – Atherosclerosis↓ | [187] | ||
AD(WD) | – NS | [187] | |||
IL-1RaTg(ic/s), apoE−/− | AD | – Atherosclerosis↓ | [190] | ||
apoE−/−, rhIL-1Ra | AD | – Atherosclerosis↓ | [191] | ||
IL-2 | IL-2r CD25 | apoE−/−, IL-2/anti-IL-2 mAb | AD | – Atherosclerosis↓ | [16] |
apoE−/−, rIL-2 | AD | – Atherosclerosis↑ | [15] | ||
apoE−/−, anti-mIL-2 | AD | – Atherosclerosis↓ | [15] | ||
IL-4 | IL-4r | IL-4−/−, ldlr−/− | AD | – Atherosclerosis↓ | [22] |
– NS | [25] | ||||
IL-4−/−, induction | AD | – Atherosclerosis↓ | [23] | ||
IL-4−/− | AD | – NS | [23] | ||
IL-4T−/− | AD | – NS | [26] | ||
IL-4−/−, apoE−/− | CC | – Atherosclerosis↓ | [24] | ||
AD/CC | – NS | [25] | |||
IL-4−/−, apoE−/−, induction | CC | – NS | [25] | ||
WT, IL-4 treatment | AD | – Atherosclerosis↓ | [27] | ||
apoE−/−, rIL-4 | AD/CC | – NS | [25] | ||
IL-5 | IL-5r | IL-5−/−, ldlr−/− | AD | – Atherosclerosis↑ | [30] |
IL-6 | IL-6r (gp130) | IL-6−/− | AD | – Atherosclerosis↑ | [37] |
IL-6−/−, apoE+/− | AD | – Atherosclerosis↑ | [38] | ||
IL-6−/−, apoE+/−, induction | AD/CC | – Atherosclerosis↑ | [38] | ||
IL-6−/−, ldlr−/− | AD/CC | – NS | [192] | ||
IL-6−/−, apoE−/− | CC | – Atherosclerosis↑ | |||
CC(16w) | – NS | [40] | |||
IL-6+/−, apoE−/− | CC(16w) | – NS | [40] | ||
apoE−/−, IL-6 lentivirus, induction | AD | – Size NS, plaque destabilization↑ | [193] | ||
apoE−/−, rIL-6 | CC | – Atherosclerosis↓ | [41] | ||
apoE−/−, rIL-6 | AD/CC | – Atherosclerosis↑ | [35] | ||
WT, rIL-6 | AD | – Atherosclerosis↑ | [35] | ||
ldlr−/−, sgp130Fc | AD | – Atherosclerosis↓ | [194] | ||
Hepatocyte-specific gp130fl/fl, apoE−/− | AD | – Atherosclerosis↓ | [36] | ||
IL-10 | IL-10r | IL-10−/− | AD | – Atherosclerosis↑ | |
IL-10−/−, ldlr−/− | AD | – Atherosclerosis↑ | [43] | ||
IL-10−/−, apoE−/− | CC | – Atherosclerosis↑ | [44] | ||
apoE−/−, mIL-10lv | AD | – Atherosclerosis↓ | [47] | ||
mIL-10Tg | AD | – Atherosclerosis↓ | [45] | ||
IL-10Tg, ldlr−/− | AD | – Atherosclerosis↓ | [48] | ||
AAV2-hIL-10, ldlr−/− | AD | – Atherosclerosis↓ | [49] | ||
AAV5-mIL-10, apoE−/− | AD | – Atherosclerosis↓ | [50] | ||
mIL-10-HVJ, apoE−/− | AD | – Atherosclerosis↓ | [51] | ||
AAV8-hIL-10, ldlr−/− | AD | – Atherosclerosis↓ | [52] | ||
IL-12 | IL-12r (β1/β2) | IL-12−/−, apoE−/− | CC | – Atherosclerosis↓ | [24] |
apoE−/−, rIL-12 | CC | – Atherosclerosis↑ | [58] | ||
ldlr−/−, IL-12 vaccine (PADRE), induction | AD | – Atherosclerosis↓ | [53] | ||
IL-13 | IL-13r | ldlr−/−, preexistent lesions, IL-13 treatment | AD | – Size NS, stable phenotype | [59] |
IL-13−/−, ldlr−/− | AD | – Atherosclerosis↑ | [59] | ||
IL-17 (A-F) | IL-17r (A-E) | IL-17a−/− | AD | – Atherosclerosis↓ | [71] |
IL-17a−/−, ldlr−/− | AD | – NS | |||
IL-17a−/−, ldlr−/−, nephrectomy | AD | – Atherosclerosis↓ | [68] | ||
IL-17a−/−, apoE−/− | AD | – Atherosclerosis↓ | |||
– Atherosclerosis↑ | [67] | ||||
– NS | [73] | ||||
Paigen | – NS | [69] | |||
CC | – NS | ||||
IL-17a−/−, apoE−/−, induction | AD | – NS | [73] | ||
IL-17a−/−, apoE−/−, induction, anti-IL-17f | AD | – NS | [73] | ||
IL-17a−/−, induction | AD | – Atherosclerosis↓ | [71] | ||
apoE−/−, anti-IL-17a ab, induction | AD | – Atherosclerosis↓ | [74] | ||
apoE−/−, anti-IL-17a ab | AD | – Atherosclerosis↓ | [74] | ||
CC | – Atherosclerosis↓ | ||||
– NS | [72] | ||||
ldlr−/−, anti-IL-17a ab | AD | – NS | [78] | ||
ldlr−/−, T-cell SOCS3 del, anti-IL-17a ab | AD | – Atherosclerosis↑ | [78] | ||
apoE−/−, Ad-IL-17RA:Fc | AD | – Atherosclerosis↓ | [76] | ||
ldlr−/−, rIL-17a | AD | – Atherosclerosis↓ | [78] | ||
apoE−/−, rIL-17a | AD | – Atherosclerosis↑ | [74] | ||
apoE−/−, rIL-17a, (IL-17a−/−) | AD | – Atherosclerosis↓ | [67] | ||
IL-17r−/−, ldlr−/− | AD | – Atherosclerosis↓ | [77] | ||
IL-17ra−/−, apoE−/− | AD | – Atherosclerosis↓ | [70] | ||
CC | – NS | [70] | |||
IL-18 | IL-18r | IL-18−/−, apoE−/− | CC | – Atherosclerosis↓ | [81] |
AD | – Atherosclerosis↑ | [195] | |||
apoE−/−, rIL-18 | CC | – Atherosclerosis↑ | [82] | ||
apoE−/−, pcDNA3-mIL18BP | CC | – Atherosclerosis↓ | [196] | ||
SCID/apoE−/−, rIL-18 | CC | – Atherosclerosis↑ | [80] | ||
SHL, pCAGGS-IL-18 | AD | – Atherosclerosis↑ | [79] | ||
CC | – NS | [79] | |||
TNF | TNFr, (p55/p75) | TNF−/− | AD | – Atherosclerosis↓ | [89] |
– NS | [96] | ||||
tmTNF | AD | – NS | [89] | ||
TNF−/−, apoE−/− | AD/CC | – Atherosclerosis↓ | |||
CC | – NS | [90] | |||
tmTNF-KI, apoE−/− | AD | – Atherosclerosis↓ | [90] | ||
CC | – NS | [90] | |||
TNF−/−, apoE*3-leiden | AD | – Advanced plaque↓, size NS | [94] | ||
apoE−/−, TNFbp | AD | – NS | [191] | ||
p55−/− | AD | – Atherosclerosis↑ | |||
p75−/− | AD | – NS | [96] | ||
p55−/−, ldlr−/− | AD | – Atherosclerosis↓ | |||
p55−/− carotid graft, apoE−/− | CC | – Atherosclerosis↓ | [100] | ||
p55−/−, apoE−/− | CC | – NS | [101] | ||
p55−/−, c. pneumonia | AD | – No effect c.pneumonia | [197] | ||
apoE−/−, chimeric murine sTNF-RI/Fc | CC | – Atherosclerosis↓ | [91] | ||
IFNγ | IFNGR | IFNγ−/−, ldlr−/− | AD | – Atherosclerosis↓ | [105] |
IFNγ−/−, apoE−/− | AD/CC | – Atherosclerosis↓ (male, female ns) | [106] | ||
apoE−/−, sIFNγr plasmid | AD | – Atherosclerosis↓ | |||
IFNγ−/−, apoE−/−, angII induction | CC | – Atherosclerosis↓ | [108] | ||
IFNγr−/−, apoE−/− | AD | – Atherosclerosis↓ | [110] | ||
apoE−/−, rIFNγ | CC | – Atherosclerosis↑ | [198] | ||
IFNβ | IFNAR | ldlr−/−, rIFNβ | AD | – Atherosclerosis↑ | [111] |
apoE−/−, IFNβ treatment | CC | – NS | [113] | ||
apoE−/−, rIFNβ, collar | AD | – Atherosclerosis↑ | [111] | ||
CC | – NS | [113] | |||
apoE−/−, IFNβ, angII (+/− collar) | CC | – Atherosclerosis↓ | [113] | ||
ldlr−/−, IFNAR1−/− myeloid BMT | AD | – Atherosclerosis↓ | [111] | ||
IFNAR−/−, apoE−/− | AD | – Atherosclerosis↓ | [199] | ||
TGFβ (1-3) | TGFβR (1-3) | AdrTGFβ | CC | – Neointima↑ | [122] |
AAV/TGFβ1 ACT, ldlr−/− | AD | – Atherosclerosis↓ | [123] | ||
apoE−/−,TGFβ1 overexpr (heart/macrophages) | AD | – Atherosclerosis↓ | |||
apoE−/−, B6-TgAlb/TGFβ | CC | – Atherosclerosis↑ | [126] | ||
apoE−/−, anti-hTGFβ1,2,3 | CC | – Atherosclerosis↑ | [116] | ||
ldlr−/−, anti-TGFβ, induction | AD | – Atherosclerosis↓ | [118] | ||
ldlr−/−, anti-TGFβ | AD | – NS | [118] | ||
TGFβRII:Fc, apoE−/− | CC | – Atherosclerosis↑ | [117] | ||
TGFβRII neg. T-cells (CD4dnTGFβRII), apoE−/− | CC | – Atherosclerosis↑ | [119] | ||
TGFβRII neg. T-cells (CD2-dnTGFβRII), ldlr−/− | AD | – Atherosclerosis↓ | [120] | ||
CD11c-dnTGFβRII, apoE−/− | CC | – Atherosclerosis↑ | [121] | ||
CD4Cre+ Smad7fl/fl, ldlr−/− | AD | – Atherosclerosis↑ | [200] | ||
IL-27 | IL-27r (IL-27ra + gp130) | IL-27−/−(Ebi3), ldlr−/− | AD | – Atherosclerosis↑ | [201] |
IL-27ra−/−, ldlr−/− | AD | – Atherosclerosis↑ | |||
ldlr−/−, rIL-27 (IL-27−/−) | AD | – Atherosclerosis↓ | [201] | ||
IL-19 | IL-20r1/2 | ldlr−/− or apoE−/−, rIL-19 | AD | – Atherosclerosis↓ | [203] |
IL-33 | IL-33r | apoE−/−, rIL-33 | AD | – Atherosclerosis↓ | [86] |
apoE−/−, sST2 | AD | – Atherosclerosis↑ | [86] |
2 The Interleukin Family
2.1 Interleukin 1
Interleukin 1 is part of the Interleukin 1 family, which consists of 11 members that have a similar gene structure, and all play a role in acute immune responses. Of these members, IL-1α, IL-1β, IL-1 receptor antagonist (IL-1Ra), IL-18, IL-33, and IL-37 are important in atherosclerosis.
Using hyperlipidemic LDL receptor (ldlr −/−) or apolipoprotein E (apoE−/−) null mice deficient in IL-1α, it was clearly shown that IL-1α exacerbates atherosclerosis [2–5]. IL-1α deficient macrophages were found to secrete lower levels of IL-1β and higher levels of IL-10 [3]. Interestingly, fatty acids, in particularly oleic acid, have been found to induce IL-1α production, further indicating a direct role of this cytokine in the pathophysiology of atherosclerosis [5].
Although the majority of studies reported a similar atherogenic effect for IL-1β [3, 6, 7], other studies found no effect on lesion size in the absence of IL-1β [2, 5]. One of these studies which has compared IL-1α and IL-1β deficient mice, found that bone marrow-derived macrophages from mice deficient in IL-1β also produce lower amounts of IL-1α, even after addition of recombinant IL-1β, suggesting that the effects seen in IL-1β-deficient mice are IL-1α-dependent [5].
The IL-1-receptor antagonist (IL-1Ra), that inhibits the effects exerted by IL-1α and IL-1β, has been extensively studied as well. Not only have murine studies conclusively shown an atheroprotective role for IL-1Ra, but also a recombinant protein of IL-1Ra, Anakinra, has been used successfully to treat rheumatoid arthritis. Recently it has been shown that the cardiovascular system in rheumatoid arthritis patients also benefits from Anakinra treatment, with a reduction in oxidative stress, endothelin and IL-6 production and improved left ventricular function [8, 9]. Currently, a clinical trial is underway investigating the effects of Anakinra treatment in patients with non-ST elevated acute coronary syndrome [10].
In innate immune cells, the NLRP3 inflammasome plays a key role in the maturation of IL-1β and IL-18 by caspase-1-dependent cleavage of their pro-forms [11, 12]. Cholesterol crystals present in atherosclerotic lesions can induce inflammation via these NLRP3 inflammasomes and thus provide an early proinflammatory factor [11]. Indeed, disabling the NLRP3 inflammasome in ldlr−/− mice markedly reduced atherosclerotic lesions size and IL-18 production thus further establishing an important role for inflammasome-associated cytokine production in atherosclerosis via interleukin 1 family members [11].
Conclusively, cytokines of the interleukin 1 family have been found to play a vital role in the initiation and propagation of atherosclerosis and could potentially provide targets for future treatment.
2.2 Interleukin 2
Interleukin 2 is required for the growth, proliferation, and differentiation of (thymic) T-cells to effector cells, and for the maturation of regulatory T-cells [13]. In humans, serum IL-2 levels correlate with carotid intima-media thickness, and thus with atherosclerosis [14]. ApoE−/− mice treated with IL-2 were found to have an increased plaque burden while mice treated with anti-IL-2 antibody featured smaller lesions [15]. Furthermore, a reduction in atherosclerosis was observed in an apoE−/− mouse model treated with an IL-2/anti-IL-2 mAb complex that was able to expand the pool of Tregs [16]. Tregs express CD25, the receptor for IL-2. When mice were treated with an anti-IL-2r (CD25) antibody, Treg development was hampered and atherosclerosis increased [17–19]. Although further research with selective targeting of IL-2 or CD25 is necessary to evaluate the net effect of IL-2 in atherosclerosis, it is clear that IL-2 plays a role in T-cell-mediated inflammation through both T helper cells and Tregs.
2.3 Interleukin 4
Interleukin 4 is secreted by T helper 2 (Th2) cells and induces T- and B-cell proliferation as well as a shift towards alternatively activated M2 macrophages, which are believed to play a protective role in atherosclerosis [20, 21]. In vascular disease, IL-4 plays an ambivalent role. Since the Th2 response is considered antiatherogenic, IL-4 was long believed to have anti-inflammatory properties. However, experimental studies have revealed a more complex role for IL-4 in the pathogenesis of atherosclerosis. IL-4 increases VCAM-1 expression in endothelial cells and increases endothelial cell turnover [20]. Furthermore, IL-4 induces the synthesis of MCP-1 and IL-6 [20], and is able to induce endothelial damage via the generation of reactive oxygen species (ROS) [20]. Accordingly, experimental atherosclerosis studies have unveiled an atherogenic role for IL-4 [22–24]. However, others have found no effects in models with genetic defects of IL-4 [25, 26], or found that IL-4 treatment induced a Th2 response and indeed decreased atherosclerosis [27]. Therefore, the role of IL-4 in atherosclerosis still remains to be fully elucidated.
2.4 Interleukin 5
Interleukin 5 is secreted by Th2 cells as well as mast cells [28]. B-cells express IL-5 receptor and IL-5 plays a role in B-cell proliferation and differentiation, immunoglobulin class switching and the production and secretion of IgM and IgA [28]. Furthermore, IL-5 induces the generation of eosinophils and drives allergic reactions [28]. In humans, a single nucleotide polymorphism (SNP) for IL-5 has been associated with cardiovascular disease [29]. In experimental studies, ldlr−/− mice receiving IL-5−/− bone marrow had increased amounts of atherosclerosis [30]. Additionally, IL-5 was found to be responsible for the generation of atheroprotective T15/EO6 antibodies by B-cells. Indeed, IL-5-knockout mice featured lower plasma levels of T15/EO6 antibodies and significantly larger lesions [30].
2.5 Interleukin 6
IL-6 and its receptor, gp130, exhibit both pro- and anti-inflammatory functions. IL-6 is secreted by a plethora of cells, including T-cells, monocytes and macrophages, but also endothelial cells, B-cells, adipocytes, fibroblasts, smooth muscle cells (SMCs) and hepatocytes [31, 32]. Once upregulated via NF-κΒ following trauma, infections or other sources of stress, IL-6 exerts its effects on a large variety of cell types. IL-6 signaling induces cell migration and proliferation, matrix metalloproteinase (MMP) production, B-cell differentiation and reduces Treg formation [31, 33, 34]. In atherosclerosis, IL-6 is produced both locally in atherosclerotic lesions as well as systemically. It can aggravate atherosclerosis through endothelial dysfunction, the recruitment as well as activation of inflammatory cells and the migration and proliferation of SMCs [31]. IL-6 can also exert effects on the receptors responsible for the uptake of LDL, the scavenger receptors SR-A and CD36 [31]. Interestingly, while these findings point towards a clear pro-inflammatory role of IL-6 in atherosclerosis, animal experiments indicate a more complex system. Studies have shown that knocking out IL-6 signaling in hepatocytes decreases atherosclerosis and that treating mice with recombinant IL-6 increases atherosclerosis [35, 36]. However, other studies showed contradictory results. For example, multiple studies found that knocking out IL-6 increases lesion size [37–40] or that recombinant IL-6 reduces lesion size [41]. These conflicting results could point towards a dual role of IL-6 in atherosclerosis although most studies indicate a predominantly antiatherogenic role for IL-6. Regardless, systemic inhibition of IL-6 seems an unlikely therapeutic method to limit atherosclerosis development, and further investigation into downstream signaling and potential site-specific effects is necessary to fully elucidate the role of IL-6 in atherosclerosis.
2.6 Interleukin 10
IL-10 is one of the most intensively studied cytokines in atherosclerosis. IL-10 is able to inhibit the presentation of antigens, block the secretion of cytokines and the expression of costimulatory molecules as well as MHC-II. It is also able to modify chemokine secretion and chemokine receptor expression [42]. IL-10 is produced by macrophages, but also by Tregs. Both knockout and overexpression strategies have revealed a protective role for IL-10 in atherosclerosis [43–52]. This makes modulation of IL-10, or its downstream modulators, a highly interesting target for future treatments.
2.7 Interleukin 12
IL-12, a cytokine consisting out of a 35-kDa light chain and a 40-kDa heavy chain, forms a bridge between the innate and adaptive immune response and promotes a Th1 based proatherogenic phenotype [53–55]. It is secreted by activated monocytes, macrophages, neutrophils and dendritic cells [56]. Furthermore, in combination with IL-18, it drives the production of IFNγ by T-cells [53, 57]. In concert with its proposed polarization towards a Th1-based response, animal studies have universally shown a pro-inflammatory and pro-atherogenic role for IL-12, as revealed in studies with IL-12 knockout mice, IL-12-treated mice and mice vaccinated against the IL-12 p40 subunit [24, 53, 58].
2.8 Interleukin 13
Similar to IL-4, IL-13 is predominantly produced by Th2 T-cells [59, 60]. Although intracellular signaling occurs via similar pathways as IL-4, IL-13 is believed to possess not just overlapping but also unique effects, including the production of TGF-β by macrophages [59, 61–63]. A recent study using ldlr−/− mice with preexisting lesions showed that treatment with IL-13 decreased plaque macrophages and adhesion molecule expression, and produced a shift from atherogenic M1 macrophages towards M2 macrophages [59]. Furthermore, IL-13−/− ldlr−/− mice fed an atherogenic diet showed significantly larger atherosclerotic lesions, indicating a protective role for IL-13 in atherosclerosis [59].
2.9 Interleukin 17
IL-17 is predominantly produced by Th17 cells, which also produce IL-21 and IL-22 [64, 65]. Th17 cells develop upon stimulation of naïve T-cells with TGFβ, IL-6, and IL-23 [66]. In recent years, it has become clear that IL-17 has an important role in atherosclerosis, although its exact effects remain elusive since several studies have reported both atherogenic and atheroprotective actions. These effects are mediated by the effects IL-17 has on both pro- and anti-inflammatory cytokines and chemokines as well as MMPs and adhesion molecules [66]. In accordance with this, studies investigating atherosclerosis in IL-17 deficient mice have yielded conflicting results regarding lesion size, including increase [67], reduction [68–71], or no effect at all [72, 73]. The use of antibodies has yielded more consistent results, with blocking antibodies reducing lesion size [72, 74–76]. Similarly, knocking out the IL-17 receptor appears to decrease lesion size [70, 77]. However, other studies using IL-17 antibodies or recombinant IL-17 also point towards a more complex, regulatory role reflecting the diverse effects IL-17 has on inflammation [67, 74, 78]. It is currently believed that the specific effects IL-17 has on atherosclerosis are dependent on the region in which IL-17 is released. IL-17 appears to have clear proatherogenic effects inside of the microenvironment of an inflammatory atherosclerotic plaque [66]. However, Th17 cells differentiating in the presence of TGF-β and IL-6 are found to produce IL-17 and IL-10, which might suppress Th1 cell differentiation [64, 66]. Conclusively, although most studies appear to point towards a proatherogenic role of IL-17, results of animal studies are inconclusive and indicate a complex, site-specific role of IL-17 and its receptors in atherosclerosis.
2.10 Interleukin 18
As discussed above, IL-18 is a member of the IL-1 family and is closely related to IL-1β, both in terms of release patterns as well as its receptors. Similar to IL-12, IL-18 has been found to be a potent inducer of the Th1 response as well as IFNγ [79–81]. The majority of animal studies point towards a proatherogenic role for IL-18. This has been validated through multiple modalities including IL-18−/− apoE−/− mice, apoE−/− mice treated with recombinant IL-18, recombinant IL-18 treatment of apoE−/− mice with severe combined immunodeficiency (SCID) and spontaneously hyperlipidemic (SHL) mice supplied with IL-18 via in vivo gene transfer, although in the latter, only mice on a high-fat diet showed a significant phenotype [79–82]. In addition to the described effects on atherosclerosis, multiple of these studies have also confirmed the importance of IFNγ in IL-18-mediated atherosclerosis. For example, the increase in atherosclerosis linked to IL-18 treatment in an apoE−/− background was nullified in mice lacking IFNγ [82]. Similarly, IL-18 knockout mice featured lower amounts of both IFNγ and Th1 cells but high Th2 counts [81]. Finally, recombinant IL-18 was found to induce IFNγ production by macrophages, NKT-cells as well as vascular cells [80]. This suggests a prominent role of IL-18 in atherosclerosis, mediated via the production of IFNγ.
2.11 Interleukin 33
IL-33 is a relatively recently discovered cytokine of the IL-1 family which is expressed in multiple cell types including endothelial cells, fibroblasts, macrophages and DCs [83]. It is believed to play a role in many auto-immune diseases and produces a Th2-based response by stimulating the production of cytokines including IL-5 and IL-13 [83–85]. Treatment of apoE−/− mice with IL-33 decreases atherosclerotic lesion size [86]. Furthermore, IL-33 increases the levels of Th2 related cytokines such as IL-4, 5, 6 and 13, as well as decreases levels of IFNγ. When IL-33 was combined with a neutralizing IL-5 antibody, which induces anti-oxLDL antibodies, the protective effect of IL-33 was nullified, indicating that IL-33 exerts its atheroprotective effects via IL-5-induced anti-oxLDL antibodies [86, 87].
3 Tumor Necrosis Factor
TNF, formerly known as TNFα, plays a role in many inflammatory diseases. Monocytes and macrophages are the main source of TNF. TNF binds two receptors, p55 and p75, of which p55 (TNFRSF1A) controls the major functions of TNF [88]. Multiple studies have shown that TNF deficiency decreases atherosclerosis in mice [89–95]. However, the role of p55 in atherosclerosis is less well established, with studies reporting both atherogenic as well as antiatherogenic effects. For example, mice only deficient in p55 were found to have increased lesion size in two studies [96, 97]. Other studies investigating mice deficient in both p55 and LDLr or apoE, however, found a decrease in lesion size [98–100], while another study found no effect [101]. The discrepancy in findings between p55 and TNF-deficient mice might be explained by p55 binding a variety of other ligands aside from TNF. The differences between various studies examining p55 are less easily explained but might be ascribed to variation in animal models. Interestingly, as observed above, mouse models featuring a deficiency in either LDLr or apoE next to p55 appeared to show different results. Perhaps the difference in homeostasis compared to mice only deficient in p55 could explain the different effects of p55 found in these studies.
4 Interferons
IFNγ plays a crucial role in immune responses and is secreted by many cells in the atherosclerotic plaque including Th1 cells, monocytes, macrophages, and NKT-cells [88, 102]. Despite its strong pro-inflammatory effects, IFNγ also exhibits anti-inflammatory characteristics and, accordingly, is able to manipulate the secretion of both pro- as well as anti-inflammatory signaling molecules [103]. In atherosclerosis, IFNγ promotes monocyte infiltration and differentiation into macrophages, foam cell formation and induces a Th1-driven response by the adaptive immune system [104]. Animal studies have confirmed this pro-inflammatory model for IFNγ in atherosclerosis. Without exception, all animal studies, using a variety of models including mice deficient in IFNγ or in IFNγ receptor as well as LDLr or apoE, show that suppressing IFNγ signaling reduces lesion size [105–110]. However, its therapeutic potential is limited due to the potential anti-inflammatory effects IFNγ has on other tissues as well as the importance of IFNγ in the immune response.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


