Clinical Pharmacokinetics and Pharmacodynamics
Basic Information 
Procedure
• ADME: Mathematical evaluation of the rates of drug absorption (A), distribution throughout the body (D), along with metabolism (M), and ultimate excretion from the body (E).
 Indicates the process whereby drug is transferred from the site of administration to the systemic circulation.
Indicates the process whereby drug is transferred from the site of administration to the systemic circulation.

 Determined by comparing the area under the plasma drug concentration curve versus time (AUC) after extravascular (EV) administration to the AUC after intravenous (IV) administration.
Determined by comparing the area under the plasma drug concentration curve versus time (AUC) after extravascular (EV) administration to the AUC after intravenous (IV) administration. 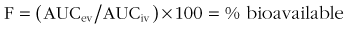

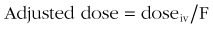










 Determined by the drug’s ability to cross biologic membranes and reach tissues outside the systemic circulation.
Determined by the drug’s ability to cross biologic membranes and reach tissues outside the systemic circulation.







 When penetration into specific tissues is unknown, a large Vd drug (Vd > plasma volume) is more likely to distribute there than a low Vd drug.
When penetration into specific tissues is unknown, a large Vd drug (Vd > plasma volume) is more likely to distribute there than a low Vd drug.
 Any condition that changes extracellular fluid volume affects the plasma concentrations of drugs with low Vd values (eg, competition with endogenous substances for protein binding sites).
Any condition that changes extracellular fluid volume affects the plasma concentrations of drugs with low Vd values (eg, competition with endogenous substances for protein binding sites).







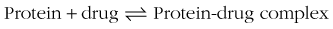






• Lipid solubility and drug ionization (the pH-partition hypothesis)










 Oral and injectable “long-acting “drug formulations are often slowly absorbed into the systemic circulation.
Oral and injectable “long-acting “drug formulations are often slowly absorbed into the systemic circulation.
 Flip-flop kinetics is identified by comparing the plasma concentration versus time curve for the extravascular route of administration to the curve after the drug is given intravenously. If the slopes of the elimination phases are not parallel, then absorption is limiting elimination (Figure 1).
Flip-flop kinetics is identified by comparing the plasma concentration versus time curve for the extravascular route of administration to the curve after the drug is given intravenously. If the slopes of the elimination phases are not parallel, then absorption is limiting elimination (Figure 1).








< div class='tao-gold-member'>

Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


