Fig. 16.1
Circadian dysfunction in locomotor activity. Mice were placed individually in cages with running wheels, and locomotor activity was recorded under different lighting conditions. Each horizontal row represents an activity record for a 24-h day. Successive days are plotted from top to bottom. The grey shading represents darkness. Mice were initially held in LD (12:12) and then released into DD. Panels show examples of the wheel-running activity recorded from WT (top) and the BACHD model of HD (bottom). The mice were 2–3 months of age
16.3.3 Disrupted Circadian Regulation of the Cardiovascular System in the BACHD Mouse Model
Poor cardiovascular health in additional to sleep-related nonmotor symptoms has been observed in HD patients. Alterations in autonomic tone that result in cardiac dysfunction is best evidenced by reductions in heart rate variability (HRV). HRV is a measure of variation in the beat-to-beat interval and reflects the dynamic balance of sympathetic and parasympathetic control of heart function and normally displays a robust diurnal and circadian rhythm. Day/night differences in HRV are lost in BACHD suggestive of a loss of circadian control over the autonomic nervous system. Additionally, an overall decrease in HRV over 24 h in BACHD mice suggests this may be due to autonomic dysfunction. Consistent with this hypothesis is the fact that heart rate and body temperature measurements during the rest phase (daytime) in BACHD mice were elevated (Fig. 16.2). Notably, a similar decrease in HRV has also been reported in HD patients beginning during the presymptomatic and early stages of HD progression (Andrich et al. 2002; Kobal et al. 2004, 2010). Reduced HRV is generally considered an indication of poor cardiovascular health and a predictor for cardiovascular disease and mortality (Bigger et al. 1992; Buccelletti et al. 2009; Thayer et al. 2010). In this regard, it is worth considering that loss of temporal control and autonomic imbalance may be promoting cardiac failure—a leading cause of death among HD patients (Chiu and Alexander 1982; Lanska et al. 1988). Electrocardiograph (ECG) parameters were also examined in WT and BACHD mice. While most parameters were not different between the two genotypes, we detected a loss of day/night differences in the PR interval, or the time it takes for the cardiac electrical impulse to travel from the sinus node of the atria to the atrioventricular node. In BACHD mice, the PR interval was significantly elongated during the active period when compared to WT mice. These findings also implicate aberrant autonomic system input to the cardiovascular system, due to its important role in regulating of the PR interval (Wallick et al. 1982; Carruthers et al. 1987). Alternatively, deficits in the sensitivity of cardiomyocyte potassium handling could affect the duration of the PR interval (Akita et al. 1998). We tested whether BACHD mice have deficits in autonomic function by examining their baroreceptor reflex, which is dependent on proper autonomic function (Schroeder et al. 2011). A blunted baroreceptor reflex response in BACHD mice suggests dysfunction of both branches of the autonomic nervous system and is consistent with significantly higher daytime blood pressure and increased HR during the rest phase in BACHD mice compared to WTs. Overall, dramatic decreases in HRV, alterations in the PR interval, and an inability to appropriately decrease heart rate and blood pressure during sleep all support the hypothesis that autonomic function is compromised in BACHD mice.
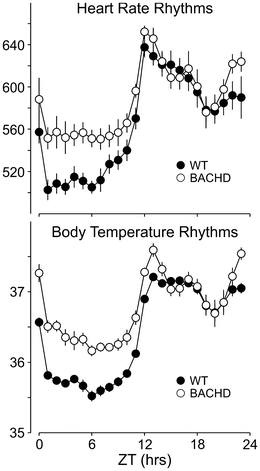
Fig. 16.2
Circadian dysfunction in physiological outputs. Examples of heart rate and body temperature measured by telemetry from WT and littermate BACHD are shown. The average waveform for each genotype as measured over 10 days in LD. The mice were 5–6 months of age
16.3.4 BACHD Molecular Clockwork May Not Be Altered
To screen whether deficits in the molecular clockwork responsible for generating circadian oscillation underlie circadian deficits observed in BACHD mice, we examined PER2 expression in the SCN at peak and trough time-points, but found no differences in BACHD mice (Kudo et al. 2011) (Fig. 16.3). These findings are in contrast to prior work using the R6/2 line that suggests behavioral impairment are accompanied by disordered circadian clock gene expression in vivo. These findings include disrupted expression of circadian clock genes centrally in the SCN and striatum, as well as peripherally in liver where core clock gene rhythms of Bmal1 and Per2 were intact but Cry1 and output gene Dbp rhythms were lost (Pallier et al. 2007; Maywood et al. 2010). Due to the conflicting findings between animal models, further work is required to definitively determine whether deficits in clock gene expression rhythms are a common phenotype in HD mouse models and, specifically, whether the BACHD line shows evidence of disruptions in its molecular clockwork beyond the SCN.
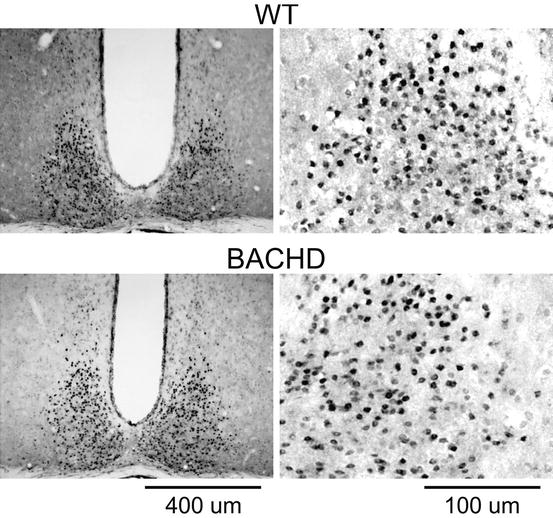
Fig. 16.3
Molecular clockwork in the SCN does not appear to be disrupted. Examples of PER2 expression in the SCN sampled from WT (top) and BACHD (bottom) mice. Mice were held in DD and wheel-running activity measured to determine circadian phase. Tissue was collected in subjective day (CT 2). IHC was used to measure PER2 immunoreactivity in the SCN. Photomicrographs of SCN tissue of each genotype in low (×10) and higher (×40) magnification. The mice were 2–3 months of age
16.3.5 Reduced Daytime Firing Rate of SCN Neurons in BACHD Mice During the Early Stages of HD Progression
SCN neurons are spontaneously active and generate action potentials with peak firing rates during the day (Colwell 2011). SCN regulates physiological rhythms throughout the brain and body using a combination of neuronal and hormonal signaling pathways. Rhythms in SCN neuronal output are believed to be necessary for the regulation of the autonomic nervous system and downstream physiological rhythms (Kalsbeek et al. 2006; Dibner et al. 2010). In the daytime, BACHD mouse SCN neurons showed significantly reduced spontaneous firing rates and an overall loss in firing rate rhythms (Kudo et al. 2011). Decreased daytime electrical activity in the SCN would therefore weaken rhythms of both neural and hormonal outputs (Fig. 16.4). This observation is consistent with the hypothesis that the weakening of electrical output from the SCN is part of the pathology responsible for the circadian behavioral phenotypes observed in the BACHD mice.
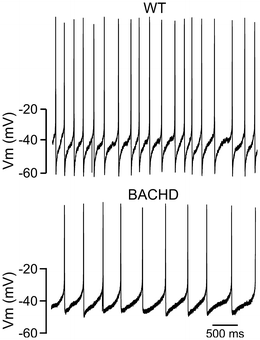
Fig. 16.4
Circadian dysfunction in neural output from SCN. Examples of spontaneous neural activity recorded from the SCN of WT (top) and BACHD (bottom) mice. The spontaneous firing rate in dorsal SCN neurons was measured during the day using the current-clamp recording technique in the cell-attached configuration. The mice were 2–3 months of age
Reduced daytime firing rates in the SCN of the BACHD line were measured from acute slice preparation of neurons embedded within a circuit. One potential mechanism by which mutant HTT may alter the function of the circadian system is by reducing the strength of intercellular coupling within the SCN circuit. Indeed, recent work in a variety of neurodevelopmental and psychiatric disorder mouse models suggests the balance between synaptic excitation and inhibition is altered and likely underlies disease-specific pathophysiology (Dani et al. 2005; Nelson and Turrigiano 2008; Gogolla et al. 2009; Milnerwood and Raymond 2010; Shepherd and Katz 2011). Inhibitory postsynaptic currents (IPSC) have been observed in cortex and striatum of R6/2 and YAC HD mouse models early in their disease progression, and these alterations are followed by late-stage decreases in spontaneous IPSC frequency in cortical pyramidal neurons (Cummings et al. 2009). Furthermore, the amplitude of spontaneous excitatory postsynaptic currents (EPSC) increases at early stages in striatal medium spiny neurons, and then in late stages the frequencies of spontaneous EPSC are decreased (Cepeda et al. 2003). These synaptic changes may underlie increased firing rates and decreased synchrony/coupling or correlated firing in both brain regions (Rebec et al. 2006; Miller et al. 2008; Walker et al. 2008). While we have no specific evidence that HD alters synaptic transmission or coupling within the SCN circuit, similar alterations in synaptic transmission is a very plausible mechanism to explain the observed disruption in circadian function in the hypothalamus.
SCN spontaneous electrical activity is controlled by a set of currents that drives daily rhythms in action potentials. During the day, SCN neurons are relatively depolarized with a resting membrane potential (−50 to −55 mV) close to the threshold for generating an action potential (−45 mV). This relatively depolarized resting potential is the result of multiple cation currents that provide excitatory drive (Pennartz et al. 1997; Jackson et al. 2004; Kononenko et al. 2004). In response to this excitatory drive, SCN neurons exhibit sustained discharge for 4–6 h without spike adaptation during their subjective day. Prior work suggests that 3 potassium (K+) currents including fast delayed rectifier (FDR), A-type-K+ current (I A), and large-conductance Ca2+ activated K+ (BK) currents are critical in the regulation of spontaneous action potential firing in SCN neurons during the day (Colwell 2011). Decreased SCN neuronal firing rates observed during the daytime in BACHD SCN may potentially be due to a reduction in magnitude of FDR and/or BK currents.
16.3.6 The Molecular Basis for Decreased Firing Rate of SCN Neurons in HD Is Unclear
It is unclear if decreased SCN electrical activity is due to changes in synaptic signaling and/or alterations of intrinsic membrane currents; however, there is a third very appealing hypothesis—disrupted mitochondrial function may also reduce neuronal electrical activity (Correia et al. 2012; Schapira 2012). Membrane repolarization following action potential generation is extremely energetically demanding. The ATP-dependent sodium–potassium pump (Na+/K+-ATPase) is critical for maintaining neuronal resting membrane potential, and this pump is more active during the day than at the night (Wang and Huang 2006). The use of this pump is very energy expensive, and insufficient ATP availability would depolarize membrane potential, leading to an inability of neurons to generate action potentials. Na+/K+-ATPase activity appear to be critical to the daily rhythm in membrane potential of SCN neurons, and therefore deficits in its activity due to insufficient ATP availability may underlie aberrant physiology in BACHD SCN.
Another mechanistic candidate underlying decreased electrical activity is mutant HTT-driven increases in oxidative damage in SCN neurons. Mutant huntingtin aggregation has been found to decrease mitochondrial efficiency by inhibition of mitochondrial trafficking (Trushina et al. 2004) and increasing reactive oxygen species (ROS) production by decreasing complex I-dependent mitochondrial respiration (Shimohata et al. 2000; Sugars et al. 2004). Notably, circadian transcriptional machinery coordinates antioxidant stress by controlling the transcription of antioxidant enzymes in many cells (Hardeland et al. 2003). Disruption of the intracellular circadian clock may thereby increase cellular susceptibility to oxidative stress and subsequently compromise function and survival (Antoch and Kondratov 2010). This hypothesis is supported by observations of disrupted ROS homeostasis in Clock mutant mice (Kondratov et al. 2006). Based on observations of disrupted clock gene expression in the R6/2 mouse model, a similar mechanism may reduce ROS homeostasis and thereby alter neuronal function (Morton et al. 2005). Unlike the R6/2 model, we did not observe deficits in circadian gene expression in BACHD mice, but we still expect to see an increase in ROS levels due to mutant HTT-related mitochondrial inefficiency. Oxidative stress damages a range of cellular processes, but due to K+ channel sensitivity to oxidative damage, it is particularly relevant to neuronal pacemaking properties (Sesti et al. 2010; Cotella et al. 2012). In the SCN, increased daytime firing rates are dependent on K+ channel currents, and therefore oxidative damage to the channels mediating these currents may underlie reduced daytime firing rates observed in the BACHD model.
Based on the literature, the most promising explanation for the decreased SCN firing frequency we observed in the BACHD model is reduced mitochondrial function and increased oxidative stress (Fig. 16.5). Future examination of these targets may provide new avenues for pharmacological intervention and in turn may provide new insights into disease progression and disease prevention.

Fig. 16.5
Circadian phenotype in BACHD mice compared to middle-aged WT mice. In our laboratory, we measured the circadian phenotype of WT mice at 12 months of age (Nakamura et al. 2011 ). We found significant reduction in activity output including reduced amplitude, coherence, and precision of wheel-running activity rhythms. Physiological outputs of heart rate and body temperature are also reduced at this age. Finally, we found that the amplitude of SCN neural activity rhythms but not PER2 expression was reduced at middle age. The phenotype of the BACHD mice at 3 months of age is similar to that seen in a middle-aged WT mouse
16.4 Future Directions
Reduced output from the SCN circuit would likely have profound consequences on patient health (Hastings et al. 2003; Takahashi et al. 2008; Bechtold et al. 2010; Reddy and O’Neill 2010). Many rhythms researchers believe that a robust SCN circuit is essential to daily and circadian rhythms of physiology and therefore good health. In recent years, a wide range of studies have demonstrated that disruption of the circadian system leads to a common cluster of symptoms, including metabolic and cardiovascular disease, as well as cognitive deficits. Many of these symptoms are also seen in HD patients. Based on the effects of circadian disruption in healthy humans and animal models, we argue that circadian dysfunction is not just a symptom of HD, but also likely to be a key part of the disease mechanism (Fig. 16.6). We have good reason to think so, because sleep/circadian disruptions occur early in the HD patients and HD mouse models. If this hypothesis is correct, circadian disruption may make the symptoms of HD worse, while stabilizing the rhythm could delay the symptoms. Recent work suggests that interventions that stabilize deteriorating daily rhythms can delay cognitive and motor symptom progression in the R6/2 line (Pallier et al. 2007). Whether this intervention improves rhythms in SCN output or is effective in other mouse models has yet to be determined.

Fig. 16.6
Speculation that circadian dysfunction could accelerate the pathology of HD. Work from several groups indicates that the mouse models of HD show a circadian phenotype. The circadian clockwork regulates mitochondrial function, reactive oxygen species homeostasis, DNA repair, and immune response. Dysfunction of this timing system is likely to contribute to chronic inflammation, mitochondrial dysfunction, and DNA damage. These processes are all thought to contribute to the pathology of HD and contribute to age-related changes in the brain. Therefore, we raise the possibility that circadian dysfunction due to genetic or environmental perturbations can accelerate the pathology of HD
Based on present understanding of the pathophysiology of the master oscillator in the BACHD mouse, future work needs to focus on understanding how mutant huntingtin alters the electrical activity within the SCN and development of interventions that can treat these disruptions. Identifying new therapeutic targets to treat sleep and circadian deficits in HD patients may improve quality of life and potentially slow disease progression for this currently incurable disease.
References
Abrahamson EE, Moore RY (2001) Suprachiasmatic nucleus in the mouse: retinal innervations, intrinsic organization and efferent projections. Brain Res 6(102):172–191
Akita M, Kuwahara M, Tsubone H et al (1998) ECG changes during furosemide-induced hypokalemia in the rat. J Electrocardiol 31(1):45–49PubMed
Albrecht U, Sun ZS, Eichele G et al (1997) A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell 91(7):1055–1064PubMed
Andrich J, Schmitz T, Saft C et al (2002) Autonomic nervous system function in Huntington’s disease. J Neurol Neurosurg Psychiatry 72(6):726–731PubMedPubMedCentral
Antle MC, Silver R (2005) Orchestrating time: arrangements of the brain circadian clock. Trends Neurosci 18(3):145–151
Antle MC, Smith VM, Sterniczuk R et al (2009) Physiological responses of the circadian clock to acute light exposure at night. Rev Endocr Metab Disord 10(4):279–291PubMed
Antoch MP, Kondratov RV (2010) Circadian proteins and genotoxic stress response. Circ Res 106(1):68–78PubMed
Aziz NA, Anguelova GV, Marinus J et al (2010a) Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat Disord 16(5):345–350PubMed
Aziz NA, Pijl H, Frolich M et al (2010b) Growth hormone and ghrelin secretion are associated with clinical severity in Huntington’s disease. Eur J Neurol 17(2):280–288PubMed
Baggs JE, Hogenesch JB (2010) Genomics and systems approaches in the mammalian circadian clock. Curr Opin Genet Dev 20(6):581–587PubMedPubMedCentral
Bechtold DA, Gibbs JE, Loudon AS (2010) Circadian dysfunction in disease. Trends Pharmacol Sci 31(5):191–198PubMed
Bigger JT Jr, Fleiss JL, Steinman RC et al (1992) Frequency domain measures of heart period variability and mortality after myocardial infarction. Circulation 85(1):164–171PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


