Fig. 1.
The RIN cannot predict the usefulness of gene expression data without prior validation. In addition, RIN values acceptable for RT-PCR may not be useable for genome-wide microarray experiments. Correlate the RIN with downstream experiments and determine the threshold RIN for meaningful results.
3 Methods for RNA Characterization
3.1 Microarrays
Gene expression profiling using microarray allows the user to obtain a relative assessment of the quantity of expressed gene products in a given tissue at a given time. This reflects an estimate of the biological environment of the cell(s) and cellular response under a given circumstance. The technology evolved out of a need to examine the complexity of biological interactions in parallel. In a single experiment, information can be obtained for thousands of genes represented on a microarray chip or array. The first microarrays contained about 1,000 transcripts or RNA targets; whereas today’s technology allows for the assessment of greater than 48,000 (this includes the >25,000 curated genes along with duplicate transcripts of genes, secondary to alternative splicing and other probes targeting unknown mRNA sequences). Over the course of 10–15 years, the technology has developed exponentially from the use of manual dot blots to robot generated complementary deoxyribonucleic acid (cDNA) chips and oligonucleotide arrays. Recently, the technique of constructing the arrays has been slightly modified to detect single nucleotide polymorphisms (SNPs) or copy number variations of genes across different samples. This technique scans a number of markers across the DNA to identify genetic variations associated with disease and is typically employed in SNP GWAS. For both of these techniques, the microarray can be custom designed to target-specific molecular pathways (e.g., targeted analysis of cancer-related genes or genes in an inflammatory pathway).
3.1.1 Basic Concepts and Platforms
The two largest companies manufacturing microarrays are Affymetrix© and Illumina©. There are a number of other microarray platforms available; however, the use of commercially developed products is encouraged to ensure robust and reproducible results. In addition, the original spotted array technology for measuring relative gene expression has been displaced by more accurate platforms, such as the Affymetrix GeneChip and the Illumina BeadChip that allows the determination of absolute gene expression levels. Gene expression microarrays generally fall into the following categories: either cDNA or oligonucleotide. Classical 3′-based microarrays were designed with probes localized only to the extreme 3′ end of the gene; these assays depended on priming the gene from the transcripts poly-A tail, which had its limitations. Most of the newer chips from Affymetrix (Gene 1.0 ST array) and Illumina (HumanHT-12 Expression BeadChip) have probes distributed across the entire length of the gene, not just the poly-A tail, providing more complete coverage. Automated sequencing detection systems, have eliminated the need for radioactive labeling by labeling hybridized cRNA with fluorescent dyes. In addition, the sensitivity of the microarray has dramatically increased over the years; for example, the sensitivity of the Illumina BeadChip is extremely high (1:250 K) since each gene is typically measured about 30 times.
cDNA arrays contain long sequences of cDNA (∼50 bases) generated from gene libraries and amplified using polymerase chain reaction (PCR), which are then printed into spotted matrices onto glass slides. Each spot corresponds to a specific gene or transcript (probe). mRNA is extracted from the cells of interest and amplified using PCR in which two types of fluorescent base pairs (Cy3 and Cy5) are inserted into the generated cDNA. These fluorescently labeled cDNAs from both cell lines are then allowed to hybridize to the glass chip with the cDNA transcripts generated from the gene libraries. The amount of hybridization to the cDNA transcripts on the glass chip is proportional to the amount of mRNA expression in the cell and can be quantified as a fold change in expression between the two fluorescent tags.
Oligonucleotide arrays (aka. oligo arrays) consist of shorter nucleotide sequences and have been pioneered by the commercial companies Affymetrix© and Illumina© (38). Since the length of the oligonucleotides is generally no longer than 25 bases, the density of these chips is much greater than that of cDNA arrays, allowing the user to assess greater numbers of gene products at the same time. Oligonucleotide arrays can be manufactured as traditional cDNA arrays, where the probes are spotted or synthesized on a two-dimensional substrate or by using BeadArray technology. Beadchips are constructed by introducing oligonucleotide bearing 3-μm beads into microwells etched into the surface of a slide-sized silicon substrate. Using the Illumina© platform, the beads self-assemble onto the beadchips resulting in an average of 30-fold redundancy of every full-length oligonucleotide. After random bead assembly, 29-mer address sequences present on each bead are used to map the array, identifying the location of each bead. Oligonucleotide sequences are selected based on their uniqueness to the target genes and may require the use of several matched sequences for high specificity to a single target. Similar to cDNA arrays, mRNA is extracted from target cells and allowed to hybridize to the oligonucleotide array. However, with oligo arrays, only a single fluorescent channel is used, thus only a single sample can be measured on one array.
3.1.2 Procedures
Procedures for microarray analyses on most commercial platforms generally contain the following and are slightly different from platform to platform: RNA amplification and cDNA synthesis, sample labeling, hybridization, wash, stain, and signal detection. The user must refer to guidelines provided by the manufacturer for specific laboratory procedures.
(a)
RNA amplification and cRNA synthesis (sample labeling):
Begin with unlabeled total mRNA to reverse transcribe to cDNA. This can be accomplished by available kits, such as the Illumina TotalPrep RNA Amplification kit to generate biotinylated, amplified RNA for hybridization to microarrays. The procedure consists of reverse transcription with an oligo (dT) primer (first strand synthesis) and a reverse transcriptase designed to produce higher yields of first strand cDNA. The cDNA undergoes a second strand synthesis and clean up to become a template for in vitro transcription. The in vitro transcription results in biotin-labeled antisense cRNA copies of each mRNA in a sample which can be hybridized to the microarray of interest.
(b)
Hybridization, wash, and signal detection:
In this step, the labeled cRNA is applied to the microarrays, hybridized overnight and washed prior to developing with a fluorophore, such as streptavidin-Cy3.
3.2 High-Throughput RNA Sequencing
High-throughput or deep sequencing is a relatively new application that can be used to characterize and quantify RNA in a massively parallel fashion. Sequencing has advantages over other methods discussed in this chapter, for example, both the estimation of alternative splicing and unbiased transcript discovery within a single experiment making it the future of profiling at both the RNA and DNA level.
Currently, high-throughput sequencing technologies are offered by only a handful of companies. Though usage of this technology is becoming widespread, it may be considered in its infancy compared to the other RNA analysis methods discussed in this chapter. Thus, protocols remain platform specific and subject to frequent revisions, concomitant with the rapid pace of innovation of next-generation sequencing. We may however discuss the methodologies, and general techniques that are common to any RNA sequencing experiment (RNAseq). A comprehensive comparison of the current competing technologies has been covered elsewhere (39–41), but put simply, the defining feature that distinguishes high-throughput sequencing from traditional Sanger sequencing is its basis in the production and assembly of vast numbers of short (26–300 nt) sequences. Choosing to utilize RNAseq over the other profiling methods discussed here yields many advantages, with the most obvious being sensitivity. Comparing RNAseq and the most common profiling technology available (microarrays) makes it clear that RNAseq has little, to no signal attributable to background noise, as the presence of a single unique sequence fragment among millions of others indicates the presence of the “parent” RNA in the original sample (42). Concomitantly, RNAseq does not have an upper limit for detection and quantification while array-based platforms may reach saturation of their probes. Nevertheless, the expense and low sample throughput of this technology will not make it suitable for all RNA profiling studies. In this chapter, we outline the key concepts involved in an RNAseq experiment.
3.2.1 Materials and Instruments
Available Platforms
To date there are five major platforms available for RNA sequencing: The Genome Analyzer (Illumina, CA), SOLiD3 (Applied Biosystems, CA), HeliScope (Helicos, MA), 454 (Roche, CT) and the open source Polonator. Each read (or sequencing tag) is short compared to conventional capillary-based sequencing. Lengths differ across the platforms, but 26–330 nt reads are typical (for a summary, see ref. (41)). Traditionally, reads as short as this would be considered a limitation, in terms of uniqueness and usefulness. This, however, is mostly absolved by the sheer weight of numbers—from hundreds of thousands to tens of millions of reads yielding massive redundancy and comprehensive coverage of most expressed transcripts. Consequently, this generates 0.5–50 Gb of data per sequencing run, which may take 2 days to greater than a week of machine run time.
Experiment Type and Sample Selection
Variations in the construction of RNA-seq libraries can generate profiles from different RNA populations within a given sample. Size fractionation of total RNA can separate miRNA, Piwi-interacting RNA (piRNA), and other small regulatory species from the majority of mRNAs and rRNAs. This can be performed by polyacrylamide gel electrophoresis (PAGE). As many of the mature forms of these RNAs have a size range of ∼19–32 nt, they may be sequenced in their entirety with most of the short read high-throughput sequencing technologies. Conversely, mRNA sequencing requires isolation of larger polyadenylated mRNA species, usually using an oligo-dT-based capture method prior to performing the sequencing protocol. While none of the reads will cover the entirety of the mRNA, due to the large number of sequencing fragments we can generate long contigs from significant overlaps between each read. It is important to note that those RNAs (i.e., many noncoding RNAs) lacking a polyA tail are excluded from sequencing experiments constructed in this manner. The primary reason for both of these selection procedures is to reduce the content of ribosomal RNA in the pool of RNAs to be sequenced, which if not removed, would make up the bulk of all sequenced fragments—a limitation of the technology as it stands. Fortunately, each sequencing platform is experiencing rapid innovation, and the fidelity and read length of the sequences is improving.
3.2.2 Procedures
(a)
Template preparation:
The first step in deep sequencing is the preparation of representative templates (a library) for each sample. The two primary methods involve either clonally amplified fragments (454, Illumina, SOLiD3) or direct sequencing of RNA (Helicos). Isolated molecules are captured and immobilized on a support surface such that they are at low enough density to remain distinct from their neighbors. The sequence of bases can be decoded by stepwise imaging of fluorescent bases. The sequencing and imaging methods are fundamentally platform specific and not covered here as they are not usually amenable to experimenter manipulation or variation.
(b)
Alignment and quantitation:
For RNAseq experiments, the sequenced fragments are normally aligned to a reference genome of interest. Your choice of reference will largely determine the level of detail that is recovered from the sequencing data. The most common option is to use a well-annotated genomic reference genome (e.g., mm9 for mouse and rn4 for rat); however, references constructed from transcriptome databases may also be used to identify known and novel RNA species. The tools for performing the alignment are usually provided along with the sequencing technology used; however, there has been considerable advancement in the commercial and open-source sectors to build aligners that rival and/or surpass those offered by the sequencing technology manufacturers. Examples are numerous (ELAND (43), SOAP (44), MAQ (45)); however, a free leading short read aligner tool set, Bowtie (46) and Tophat (47), provides a rapid “desktop” computing approach for aligning high-throughput sequence data to mammalian genomes, which due to their size, presents challenges beyond those of other experimental systems. From this analysis, we can observe the several advantages of RNAseq over other profiling technologies, such as microarrays and Northern blotting, namely new exon discovery, splice variation and alternate 5′ and 3′ transcription start and stop sites (Fig. 2).
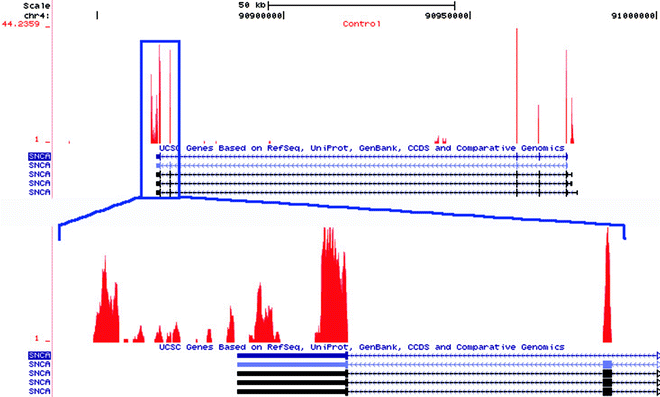
Fig. 2.
Distribution of RNAseq tags from an RNAseq experiment on human entorhinal cortex, after alignment with Bowtie across the Synuclein gene (SNCA). The upper panel shows peaks of sequence tags across all of the exons of SNCA with some evidence of transcriptional activity originating from intron 4. Boxed area shows zoomed view of the 3′UTR of SNCA where the considerable density of observed sequence tags likely indicates an alternate 3′ termination site than from that annotated.
Apart from transcript discovery and identification, the primary goal of RNAseq experiments is to quantitate the number of sequences found across samples. As in the early years of microarray development, there is no clear consensus on the optimal method for normalizing RNAseq data for comparison across samples. For a review of the current statistical tools being utilized, see Bullard et al. (48). It is likely that variations of scaling the data, such as calculating the reads per kilobase of exon model per million mapped reads (RPKM) will prove to be the most robust. Tools for quantitative analysis are less abundant than those for alignment, but commercially available suites, such as CLC Genomcis work bench (CLC Bio, Denmark) and Lasergene (DNASTAR, UK), are accessible options for non-bioinformaticians.
3.3 Northen Blotting
Though it has been superseded in some aspects by other RNA analysis techniques discussed in this chapter, Northern analysis remains a powerful method for quantification and characterization of transcription. We may still apply Northern analysis to obtain a relative comparison of abundance between samples on a single support and it remains the simplest method for determination of transcript size and hence, detection of alternate splicing.
One of the strengths in performing a Northern blot is in its relative simplicity and low cost compared to other RNA characterization methods, making it accessible to most laboratories (49, 50). Purified RNA samples are separated by size via denaturing gel electrophoresis, and then the RNA is transferred to a membrane, cross-linked, and probed for the gene of interest with a labeled nucleic acid. The procedure is flexible and customizable to your particular laboratory (e.g., Probes may be anything from p32/p33 radiolabeled DNA, non-radiolabeled DNA, RNA, or oligonucleotides may all be used).
As with other RNA characterization methods, selecting the correct assay for your experimental paradigm requires consideration of the limitations of each system. Northern blots are particularly sensitive to RNA degradation (51). If the integrity of the RNA is not maintained (Clear 28S Ribosomal bands of double intensity of the 18S), then the signal from many probes may be reduced or lost. As with all the techniques throughout this chapter, RNase-free reagents and techniques are required for each step. Additionally, Northern blots generally require substantial amounts of RNA (5–50 μg or greater); quantities not always generated depending on the brain region or blood sample being examined. Lower quantities of starting RNA generally yield less sensitive results depending on the abundance of the transcript being probed, but in almost all cases Northerns will be less sensitive than qRT-PCR (52).
3.3.1 Materials and Reagents
(a)
10× MOPS solution:
50 mM anhydrous sodium acetate.
10 mM EDTA.
0.2 M MOPS.
Bring volume to 700 ml with DEPC-treated water and adjust the pH to 6.5. Bring to final volume of 1 L.
(b)
RNA sample buffer (1 ml):
500 μl deionized formamide.
180 μl formaldehyde (37%).
216 μl DEPC water.
100 μl of 10× MOPS.
4 μl EtBr (from stock of 10 mg/ml).
(c)
10× Loading dye:
0.25% Bromophenol blue.
0.25% Xylene cyanol.
20% Ficoll.
Dissolve in DEPC water. Alternately, any commercially available RNase-free loading dye may be used, provided the migration rate is known.
(d)
20× SSPE:
175.3 g of NaCl.
27.6 g of NaH2PO4 × H2O.
7.4 g of EDTA.
Adjust pH to 7.4 in a 700 ml volume then and bring to final volume of 1 L.
3.3.2 Procedures
1.
Denaturing agarose-formaldehyde gel electrophoresis of RNA:
(a)
Prepare a 1% agarose gel. For 250 ml, dissolve 2.5 g agarose with 25 ml of 10× MOPS and 205 ml of water. When the flask has cooled, add 12 ml formaldehyde. Pour the gel and allow it to set at room temperature. Place the gel in a gel tank and add 1× MOPS running buffer. Note: Formaldehyde is toxic and casting should be carried out in a fume hood and the gel should be covered when in use. Cast the gel as thin as practical as this will enhance transfer of high molecular weight RNA to the membrane.
(b)
Load samples onto the gel. During the final step of RNA extraction samples should be resuspended in at least 12 μl of sample buffer. Heat samples for 5 min at 65°C, place on ice, and then add 3 μl of loading buffer.
(c)
Run the gel at 100–200 V. The length of time will vary depending on the length of your gel, but a general rule is to run until the bromophenol blue has migrated two-thirds of the length. Image the gel with a UV transilluminator and document in the presence of a fluorescent ruler. Place gel in 20× SSPE while preparing the next step.
2.
Capillary transfer of RNA to a membrane support:
(a)
Fill the reservoirs of an electrophoresis tank with 20× SSPE. Cut a wick from a piece of Whatman 3MM paper long enough to span both reservoirs. Cut four pieces of Whatman 3MM paper the size of the gel, wet with 20× SSPE, and place two in the center of the tank on top of the wick. Place the gel on the filter paper and roll out air bubbles. Cut four strips of parafilm and place under each edge of the gel. Cut a piece of positively charged nylon membrane (Ambion) the dimensions of the gel and position on the surface of the gel. Place the remaining two pieces of Whatman 3MM paper on top of the membrane, roll out any air bubbles. Stack paper towels on top of the Whatman 3MM paper to a height of approximately 10 cm, and add a light weight. Leave to transfer overnight. Faster commercially available transfer methods, such as vacuum and electroblotting can also be used.
(b)
Rinse the membrane with 5× SSPE and then allow to dry. Place in a UV cross-linker to bind the RNA to the membrane. Alternately, the membrane may be baked at 80°C for 2 h.
3.
Get Clinical Tree app for offline access
Hybridization of labeled probes:
(a)




Prepare labeled probes specific to your requirements (e.g., Prime-It® II Random Primer Labeling Kit, Stratagene). Prehybridize the membrane in 5 ml of hybridization solution (5× SSPE, 50% (w/v), 5× Denhardt’s solution, 1% SDS, 10% Dextran Sulfate Na salt). Incubate with rotation for 1–3 h at 42°C for DNA probes (60°C for RNA probes).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
