Fig. 1
Cellular responses following the opening of transient receptor potential melastatin 2 (TRPM2) cation channels and the associated increase in intracellular calcium concentration. The channels are containing C (Nudix box) and N sections. The Nudix box domain has ADP-ribose pyrophosphatase activity. The TRPM2 channels can be gated by H2O2 and ADP-ribose via activation ADP-ribose pyrophosphatase
Table 1
Role of inhibitor and gate in oxidative stress and ADPR on TRPM channels in different neuronal cells
Activator | Cells | Effects | Reference |
|---|---|---|---|
ADPR | Rat microglia | Activator unknown | |
Oxidative stress | Striatal neurons | Activator | |
C13 microglia cells | Activator | [9] | |
Both ADPR andoxidative stress | Cortical neurons | Activator | [19] |
No effects | [21] | ||
ADPR | Pyramidal neurons and | Insufficient | [41] |
CA1 interneurons of hippocampus | |||
Oxidative stress | Pyramidal neurons and CA1interneuron of hippocampus | Activator | [41] |
Oxidative stress | Rat primary striatal cultures | Activator | [8] |
Oxidative stress | Astroglia | Activator | [2] |
TRP Superfamily
The transient receptor potential (TRP) cation channel superfamily compromises a diverse range of voltage-dependent Ca2+-permeable cation channels. There are 28 mammalian TRP channels, grouped into seven subfamilies. All members of the TRP channel superfamily, which includes the TRP cononcial (TRPC) subfamily consisting of seven, the TRP vanilloid (TRPV) subfamily consisting of six, TRP melastatin (TRPM) subfamily consisting of eight, TRP polycystein (TRPP) subfamily consisting of three, TRP mucolipin (ML) subfamily consisting of three, and the TRP ankyrin (TRPA) subfamily consisting of one are poorly characterized, but are attracting increasing interest because of their involvement in several human diseases. TRP channels have a basic structure similar to voltage-gated potassium channels, with homo- or heterotetrameric arrangements around a central ion conducting pore between the fifth and sixth segments of the channel pore [5]; the fourth transmembrane segment is not positively charged. The N-termini of TRPV and TRPC, but not those of TRPM channels, contain multiple ankyrin binding repeats. The C-terminal part of the sixth segment in TRPC and TRPM channels includes the “TRP domain,” a conserved stretch of 25 amino acids starting with the nearly invariant “TRP box” that is missing in TRPV channels. In addition, all TRP channels have multiple regulatory protein interaction sites. Multiple protein kinase A (PKA) and C (PKC) putative phosphorylation sites have been identified and partially tested for function. Phosphatidylinositide 3-kinase SH2-recognition domains have also been identified in several TRP channels [53].
TRP channels contribute to changes in cytosolic free Ca2+ [Ca2+]i by acting as Ca2+ entry channels in the plasma membrane directly or by changing membrane potentials, modulating the driving forces for the Ca2+ entry channels. All functionally characterized TRP channels are permeable to Ca2+ with the exceptions of TRPM4 and TRPM5, which are only permeable to monovalent cations but not to Ca2+ or Mg2+ (see Fig. 2). Two mammalian TRPs, TRPV5 and TRPV6, are highly Ca2+ permeable. TRPM6 and TRPM7 are highly permeable to Mg2+. At least three TRP channels, TRPV1, TRPML1, and TRPP3, are highly permeable to H+ ions [38].
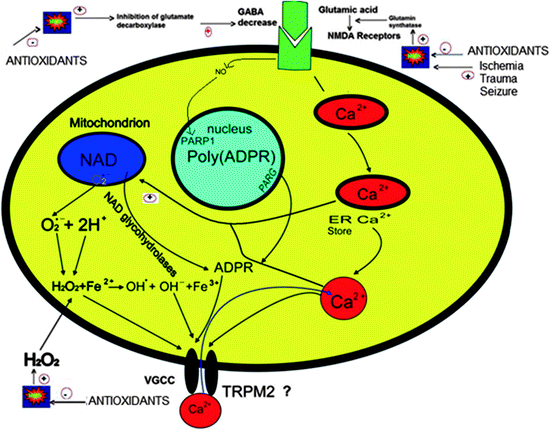
Fig. 2
Cells regulate intracellular Ca2+ levels lightly and excessive Ca2+ loads can lead to inappropriate activation of processes that normally operate at low levels, causing metabolic derangements and eventual cell death. For example, excess elevations in intracellular Ca2+ may activate enzymatic degradation, induce formation of reactive oxygen species, or disrupt normal mitochondrial function leading to oxidative stress and bioenergetic failure. Excessive Ca2+ load, in particular via N-methyl-d-aspartate (NMDA) receptors (NMDAR), is toxic to neurons in neurodegenerative diseases. NMDAR-mediated Ca2+ entry triggers a neurotoxic signal cascade involving the activation of neuronal nitric oxide (NO) synthase (nNOS), formation of toxic reactive oxygen species (ROS) and NO, and activation of the proapoptotic protein poly(ADP-ribose) polymerase (PARP-1). Antioxidants regulate Ca2+ influx into cytosol by inhibition of ROS. Sustained depolarization of mitochondrial membranes and enhanced ROS production activate transient receptor potential melastatin 2 (TRPM2), voltage-gated Ca2+ channels (VGCC), and Ca2+ influx increases by activation of TRPM2 via ROS. The molecular pathway may be a cause of neurological symptoms and thus represents a fruitful subject for further study
TRPM
The TRPM2 subfamily consists of eight members, subdivided into three subgroups on the basis of sequence homology: TRPM1/TRPM3, TRPM4/TRPM5, and TRPM6/7, with TRPM2/TRPM8 being distinct proteins ([31]; Fig. 3).
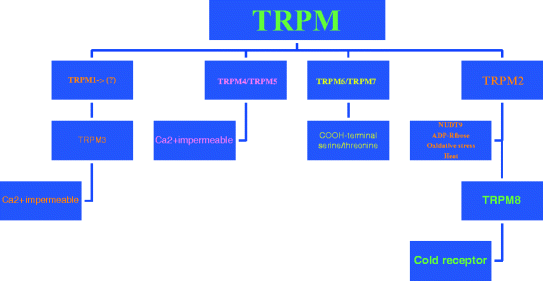
Fig. 3
The transient receptor potential melastatin 2 (TRPM2) subfamily consists of eight members, subdivided into three subgroups on the basis of sequence homology: TRPM1/TRPM3, TRPM4/TRPM5, and TRPM6/7, with TRPM2/TRPM8 being distinct proteins
Three members of this subfamily carry an entire functional enzyme in their COOH-termini: TRPM2 contains a functional NUDT9 homology domain, exhibiting ADPR pyrophophatase activity (Fig. 1), whereas both TRPM6 and TRPM7 contain a functional COOH-terminal serine/threonine kinase. Except for TRPM1 which has not been functionally characterized until now, all TRPM channels are cation channels, although the Ca2+ permeability is diverse, ranging from highly permeable (TRPM6/TRPM7 and splice variants of TRPM3) to Ca2+ impermeable (TRPM4 and TRPM5). TRPM4 and TRPM5 are heat-sensitive, Ca2+ activated channels [39]. TRPM2 is activated ADPR, H2O2, and heat. TRPM3 channels are, much like TRPM6 and TRPM7, regulated intracellular Mg2+ levels. They show considerable constitutive activity although TRPM3 activation was also reported after cell swelling and by sphingosine. TRPM8 is the infamous cold receptor [6, 13, 37, 39, 56].
TRPM2 Channels
One of the TRP channels is TRPM, so named because the founding member, TRPM1, was first described in connection with metastatic melanomas [6]. TRPM2 was formerly known as TRPC2 and LTRPC2. TRPM2 channels were first identified in 1998 and subsequently recognized as members of the TRPM family [5]. All eight TRPM family members have been linked to disease, either by functional studies in mouse models or by genetic evidence [39].
Molecular Mechanisms in the Activation of TRPM2
Oxidative Stress
ROS including superoxide anion, hydrogen peroxide (H2O2), and singlet oxygen act as subcellular messengers in such complex processes as mitogenic signal transduction, gene expression, and regulation of cell proliferation when they are generated excessively or when enzymatic and nonenzymatic defense systems are impaired [31, 32]. Although many intra- and extracellular molecules may participate in neuronal injury and cell apoptosis, accumulation of oxidative stress due to excessive generation of ROS appears to be a potential factor for cell damage and death [12].
Oxidative stress for which application of H2O2 is an experimental paradigm, induces TRPM2 currents and an increase in free intracellular Ca2+ [Ca2+] in various cell types transfected with TRPM2 [36], as well as in pancreatic β-cells [18], neutrophil granulocytes [14], and U937 monocytes [44, 56]. However, the exact mediator molecules for H2O2-induced TRPM2 channel activation remain to be identified. The ROS sensitivity of TRPM2 can be mediated by NAD+ [13] or ADPR [54] released from mitochondria or through protein oxidation [11]. H2O2 and cADPR have been proposed to potentiate the effect of ADPR at lower concentrations and to gate the TRPM2 channel directly at higher concentrations [27].
Role of Oxidative Stress and ADP-Ribose /NAD Productsin the Activation of TRPM2
Ca2+ influx via TRPM2 is thought to occur via production of ADP ribose (ADPR). ADPR may arise from a mitochondrial source or alternatively via activation of poly(ADPR). ADPR may also arise from a mitochondrial source [11].
NAD+ has been reported to stimulate TRPM2 [13]. TRPM2 contains a characteristic structural feature known as a Nudix domain in its C-terminal cytosolic tail [44]. A Nudix domain is a consensus region that is known to be present in a class of pyrophosphatases that degrade nucleoside diphosphates ([31]; Fig. 1). Indeed, the Nudix domain of TRPM2 cleaves ADPR, a breakdown product of NAD+ and cyclic ADPR, representing an intracellular second messenger that stimulates calcium release mediated by ryanodine receptors [33]. ADPR is hydrolyzed by TRPM2, however, it also activates TRPM2 and induces TRPM2 currents during infusion of ADPR by the patch pipette [13, 44]. NAD+ has been reported to stimulate TRPM2 [13, 23, 25]. In our recent study, we observed the activation of TRPM2 by NAD+, which supports the idea that this substance activates TRPM2 directly [35, 36].
Previously published data indicated that TRPM2 can be gated by H2O2 and it has been suggested to gate the channel independently of ADPR [13, 54]. Because Perraud et al. [44] demonstrated that reducing the ADPR concentration within mitochondria largely suppresses H2O2-mediated activation of TRPM2, the gating mechanism of H2O2 is primarily based on its ability to release ADPR from mitochondria, which then proceeds to activate TRPM2. However, Kolisek et al. [20] recently demonstrated that H2O2 may be a self-sufficient stimulus for TRPM2 activation, as it can initiate the release of ADPR from mitochondria and at the same time function as a potentiating cofactor of ADPR. In our recent study, we observed that the H2O2-mediated activation of TRPM2 appears to result from a direct gating mechanism, because TRPM2 activation by H2O2 was relatively rapid in whole cell recordings, and the compound triggered single-channel activity in excised membrane patches [35]. After adding H2O2 to the bath, cell inside-out records were washed by intracellular buffer and then H2O2 was given again. The channel was activated again by a second administration of H2O2 although no intracellular component was present. From, this we concluded that H2O2-induced, single-channel activity observed in excised membrane patches is likely caused by a direct gating mechanism.
Methodological problems have led to different reports on the activation of TRPM2 by NAD+, ADPR, and H2O2. For example, in a previous study [54] using HEK293 cells as an expression system, we demonstrated characteristic currents through the splice variant of TRPM-ΔC induced by H2O2. In later experiments we could also stimulate them with NAD+, although stimulation with H2O2 was not possible in the CHO cell expression system using wild-type TRPM2 with a sufficient consistency [20].
A second problem with different results arises from the use of different cell types to study TRPM channel activation by H2O2, ADPR, and its metabolites. On the other hand, the channel activation by H2O2 appears to represent a cell-specific process in cells with endogenous expression of TRPM2. For example, the TRPM2 channel is activated by H2O2 in HEK293 cells [13, 54], CHO cells [37], CRI-GI rat insulinoma cell lines [18], and rat primary striatal cultures [8], but not in human neutrophil granulocytes [14]. Kolisek et al. [20] used cADPR and the compound stimulated TRPM2 in HEK293 cells; that channel was opened by cADPR. Later, the report was supported by the results of Gasser et al. [10] and TRPM2 channels in Jurkat cells were opened by cADPR. However, a more recent study did not support the results for neutrophil granulocytes [15] and TRPM2 channels did not open by stimulation with cADPR.
Role of TRPM2 Channels in Neurological Cells
Northern blotting and quantitative PCR techniques indicated that TRPM2 is broadly expressed in the CNS. However, as this evidence was derived from homogenized tissue samples, it does not allow expression in neurons to be distinguished from that in glia. The importance of making such a distinction is highlighted by a recent study that failed to identify TRPM2 transcripts or functional channels in cerebellar granule cells and astrocytes [21]. Rather, TRPM2 was detected in microglial cells leading to the suggestion that the CNS distribution of TRPM2 corresponds to its expression in nonneuronal cells [22, 41, 45].
Role of TRPM2 Channels and Oxidative Stressin Microglia and Astroglia Cells
The NADPH-oxidase in phagocytic cells such as microglia is an electron transport system that catalyzes the reduction of oxygen to superoxide radical. Under physiological conditions, the system contributes to the elimination of pathogens but under chronic inflammatory conditions such as scleroderma or liver fibrosis it is thought to induce neurodegeneration by the massive accumulation of superoxide radicals. Monocyte superoxide, in high glucose media, is released by the NADPH-oxidase but not by the mitochondrial respiratory chain, and antioxidants such as α-tocopherol inhibits superoxide release via inhibition of protein kinase C (PKC)-a. The assembly of a functional NADPH-oxidase complex at the plasma membrane depends on the phosphorylation and subsequent translocation of several cytosolic subunits (p40phox, p47phox, p67phox, and Rac1/2; [4]). In microglia cells, antioxidants inactivate PKC via the phosphotase-mediated pathway (PP1 or PP2A) and, as a consequence, blocks the phosphorylation-dependent translocation of p67phox to the plasma membrane. As a result, the production of superoxide radical by the microglial NADPH-oxidase system is substantially inhibited, offering a partial explanation for the beneficial effect of antioxidants such as α-tocopherol on a variety of neurodegenerative diseases [31].
Like other activated macrophages, microglia remove bacteria and cellular debris and produce a diverse range of mediators of the inflammatory cascade including arachidonic acid derivates and H2O2. Thus microglia cells are a key factor in the immune defense and tissue repair in the CNS. Kraft et al. [21] described that novel calcium influx pathway in microglia cells coupled to hydrogen peroxide and ADPR and provided evidence that this pathway involved TRPM2 although they failed to detect TRPM2 in cultured cerebellar granule neurons. Recently, Ohana et al. [40] investigated expression of putative Ca2+-permeable TRPM2 channels in rat-cultured microglia cells by quantitative real-time RT-PCR. They detected transcripts in the rat-cultured microglia cells for TRPM2 genes.
Although the role of astroglia in the progression of neurodegenerative disease is still relatively unknown, their importance in regulating the normal and abnormal neuronal environment is attracting increasing attention. Through Ca2+ signaling cascades, astroglia control gene expression, neuronal differentiation, and programmed cell death, which are all integral to developmental and degenerative processes. Under conditions of oxidative stress, glial cells provide energy support for neurons, exert a protective function by scavenging and detoxifying ROS, and direct neuronal resistance or vulnerability to degeneration through Ca2+-dependent secretion of trophic or inflammatory factors [30]. There are few reports on TRPM2 in astroglia cells. Bond and Greenfield [2] reported that the additive effects of L-VGCC blockade and TRPM2 inhibition during oxidative stress significantly enhanced recovery from protein synthesis suppression and repressed subsequent compensatory protein overexpression. These results indicated that Ca2+ signaling is integral to astroglial transcriptional and translational responses to oxidative stress.
TRPM2 Channel in Hippocampal Neurons
TRPM2 is expressed in diverse cell types and despite convincing evidence for high expression in the mammalian brain, much of this signal is attributed to strong expression in nonneuronal cells. Thus, the existence of functional TRPM2 channels in neurons is controversial at best. Recently, Olah et al. [41] reported that functional TRPM2 channels are highly expressed in pyramidal hippocampus neurons, including those of CA1 interneurons in hippocampal slices. They also reported that ADPR alone is insufficient to gate TRPM2 in hippocampal neurons. They concluded that concomitant influx of Ca2+ through voltage-dependent Ca2+ channels and/or NMDAR is necessary to fully activate TRPM2 channels.
TRPM2 Channels in Dorsal Rood Ganglion Cells
There are several types of sensory neurons in the dorsal rood ganglion (DRG), with responsiveness to different external and internal stimuli. These stimuli, including nociceptive, thermal, or mechanical, activate different receptors and ion channels that are present in the nerve terminals at the sensory receptive fields and their expression in selective subsets of DRG neurons determines the response profile of nonselective cation channels that play important functions in sensory neurons. TRPM8 is the only TRPM channel with a clearly assigned function in DRG neurons. It is activated by innocuous cool stimuli and responds to menthol and icilin with intracellular Ca2+ elevations [43]. Recently, TRPM2 channels were firstly detected in DRG mouse cells [50]. They also observed that levels of TRPM2 channels in DRG cells were significantly higher in lumbar tissue than in thoracic tissue in the adult mouse although they did not observe the channels in the DRG cells of embryonic day 12–12 weeks of age.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


