CHAPTER 10 Cardiovascular Diseases
Cardiovascular (CV) system function is critical to exercise, thermoregulation, and the blood flow–dependent functions of the lungs, kidneys, gut, and reproductive system. Heart and vascular diseases are common in horses, but fortunately the underlying lesion is often minor and well tolerated. However, clinically significant CV disease can develop in horses with clinical signs that can include arrhythmia, exercise intolerance, congestive heart failure (CHF), weakness or collapse, systemic infection, or sudden death.
No recent comprehensive studies of CV disease prevalence are available. Holmes, Darke, and Else33 observed that 2.5% of hospitalized horses were in atrial fibrillation (AF).33 Else and Holmes27–29 noted myocardial fibrosis in 14.3% of horses examined at necropsy and evidence of chronic valvular disease in approximately 25% of the hearts examined. Various CV lesions were considered important in 8.5% of 480 consecutive losses in a necropsy study conducted by Baker and Ellis.52 CV diseases are probably the third most common cause of poor performance after musculoskeletal and respiratory diseases.53–56 Occult heart disease, cardiac arrhythmias, and vascular lesions are considered important reasons for unexplained sudden death.52,57–69 Certainly, most clinicians encounter manifestations of CV disease or dysfunction on a regular basis.
The assessment of CV disease in a horse is predicated on a competent clinical examination, the clinician’s knowledge, and the ability to order and evaluate diagnostic studies. Incomplete information may impede an accurate diagnosis, foster miscommunication of risks to the client, or delay the proper course of management. Fundamentals of CV anatomy, physiology, and electrophysiology are reviewed elsewhere.9,20,70–77 This chapter offers a framework for understanding the lesions, pathophysiology, diagnosis, and management of important congenital and acquired conditions of the heart and major vessels. Clinical aspects of circulatory shock are described in this volume, and the reader is referred elsewhere for the management of cardiopulmonary arrest.78,79
ANATOMIC CORRELATES OF CARDIOVASCULAR DISEASES
Most diagnostic techniques, including cardiac auscultation, electrocardiography, cardiac catheterization, and echocardiography, are predicated on an understanding of cardiac anatomy and physiology. The CV system is divided into two separate circulations: (1) systemic and (2) pulmonary. The systemic circulation has a greater venous capacitance, ventricular pumping pressure, arterial pressure, and vascular resistance.73,75–77 Despite these differences, the functions of these two circulations are interdependent, as the following examples illustrate. Systemic and pulmonary circulations are arranged in series, therefore cardiac output (CO) from the left ventricle (LV) and the right ventricle (RV) must be equivalent. Accordingly, failure of either ventricle limits CO. Isolated LV failure, as with severe mitral regurgitation (MR), can cause right-sided failure; this is explained by the increased pulmonary venous pressure causing pulmonary hypertension and imparting a pressure load on the RV. A third example is the case of isolated RV failure with marked RV dilatation. Here the leftward bulging of the ventricular septum impairs filling of the LV. This last situation also can develop in chronic pericarditis. Arrhythmias also affect both sides of the heart so that the development of AF in the setting of structural heart disease can promotes biventricular heart failure.
The heart consists of unique active and passive components,75 and different diagnostic methods are needed to evaluate these structures and associated functions. Normal heart action requires coordination of electrical activity, muscular contraction and relaxation, and valve motion. When reviewing heart anatomy, and subsequently cardiac pathology, it is useful to consider the anatomic integrity of the pericardium, myocardium, endocardium, and valves, specialized impulse-forming and conduction systems, and blood vessels.70 Using this approach, the causes of CV disease can be conveniently subdivided into anatomic, physiologic (functional), and causative diagnoses (Boxes 10-1 and 10-2).
BOX 10-2 CAUSES OF CARDIOVASCULAR DISEASE
Congenital Cardiac Malformation
Pericardial Disease
Pericardial effusion can develop as a primary disorder or secondary to pleuropneumonia. Infective pericarditis can produce an effusion sufficient to cause cardiac tamponade or eventual constriction of the heart.55,80–97 Sterile, idiopathic pericardial effusion also has been reported in horses. The volume of effusion can be substantial and can lead to cardiac decompensation.87 Cardiac mass lesions and intrapericardial tumors have been reported sporadically.89,92,98,99 Cranial mediastinal tumors (lymphosarcoma) or abscesses secondary to pleuropneumonia also can compress the heart and mimic pericardial disease.100 Clinical aspects of pericardial disease are discussed later in this chapter.
Myocardial Disease
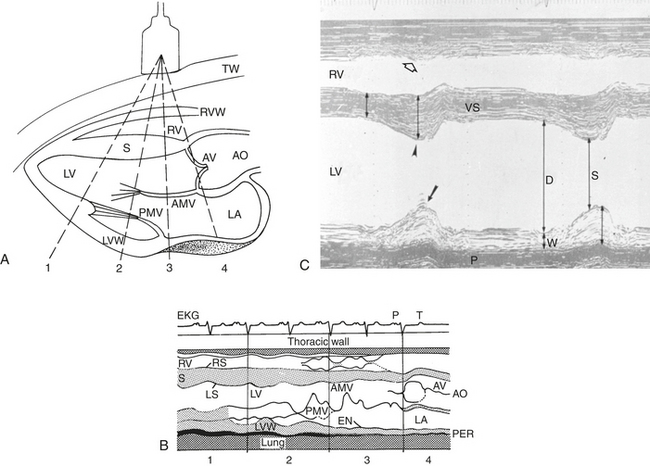
The myocardium forms the bulk of the atrial and ventricular muscular walls. The right atrium (RA) communicates with the RV inlet through the right atrioventricular (AV) or tricuspid valve. The RV appears crescent shaped on cross-sectional echocardiographic examination and is functionally U shaped. The RV inlet is located in the right hemithorax and the outlet, pulmonary valve, and main pulmonary artery (PA) on the left side of the chest. The left atrium (LA) is caudal to the RA and separated by the atrial septum. The LA is dorsal to the LV through which it communicates across the left atrioventricular (mitral) valve. The LV is circular in cross-section when viewed by echocardiography and separated from the RV by the ventricular septum. The septum and free walls are thicker than the RV free wall (by approximately 2.5 to 3 times). Persistent embryologic openings in the cardiac septa are known as septal defects, with the ventricular septal defect (VSD) representing the most common cardiac anomaly in most equine practices (see Box 10-2). The LV is functionally V shaped with an inlet and outlet separated by the cranioventral (septal or “anterior”) leaflet of the mitral valve (Figure 10-1). The aorta originates in the LV outlet, continuous with the ventricular septum cranially and in fibrous continuity with the septal mitral leaflet caudally. This great vessel exits from near the center of the heart and to the right of the main PA.
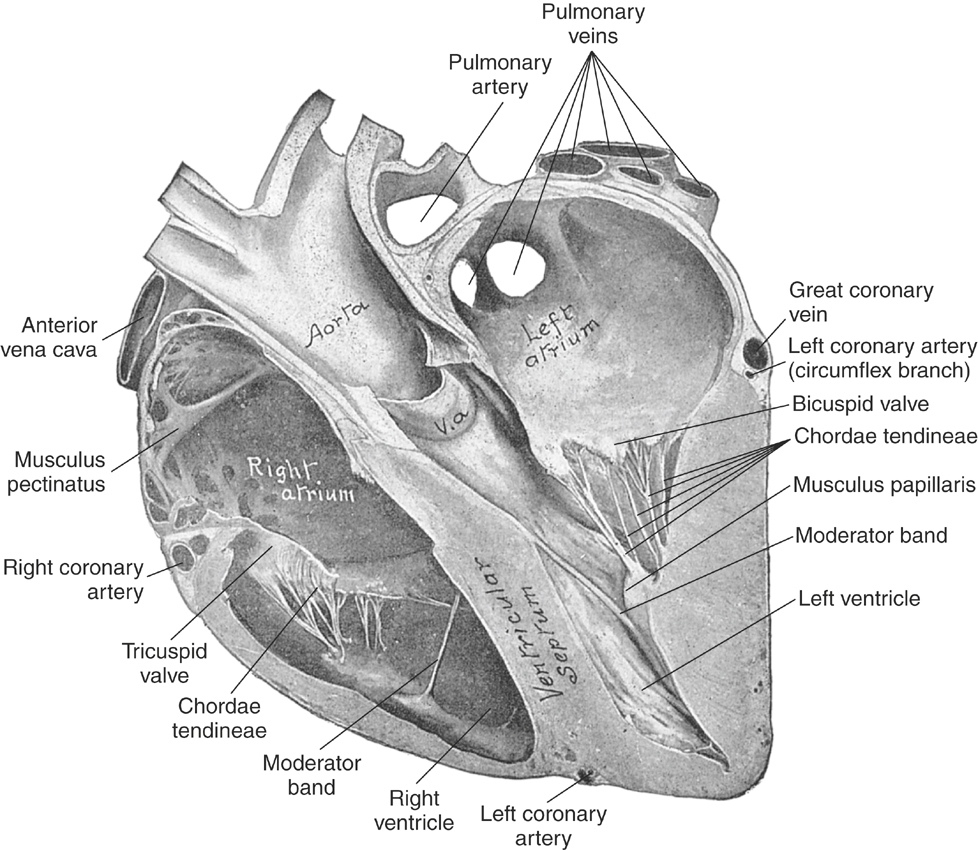
The myocardium may dilate or hypertrophy in response to exercise,101,102 increased work caused by structural cardiac disease, or as a consequence of a noncardiac disorder. Ventricular or atrial dilatation is recognized echocardiographically or at necropsy by distention and rounding of the affected chambers, including a “double-apex” sign when marked RV enlargement occurs. Lesions causing systolic pressure overload lead to concentric hypertrophy.103 More common in horses are lesions such as incompetent valves or shunts that cause ventricular volume overload with dilatation and eccentric ventricular hypertrophy. Increased cardiac work also occurs in response to exercise, severe anemia, and infections. In these situations, compensatory increases in CO, sympathetic activation, and peripheral vasodilation occur to maintain oxygen delivery to the tissues.104,105
The overall prevalence of myocardial disease is unknown; however, multifocal areas of fibrosis are commonly found at necropsy.28,29,106–110 Whether these areas indicate prior inflammation, toxic injury, or ischemic necrosis caused by intramural coronary disease is uncertain. Cases of multifocal or diffuse myocarditis have been observed. Myocardial inflammation and myocardial failure can lead to cardiac arrhythmias and heart failure.4,111,112 Idiopathic dilated cardiomyopathy develops sporadically and is recognized echocardiographically or by nuclear studies as a dilated, hypokinetic LV or RV.90,113 Ingestion of monensin or other ionophores can cause mild to severe myocardial injury.114–120 Neoplastic infiltration is considered rare.59,98,99,121 Impaired myocardial function as a consequence of regional ischemia has been sought using stress echocardiography immediately after treadmill exercise or pharmacologic stress, but this diagnosis requires further definition.122 Myocardial contraction is dictated by electrical activity of the myocardium; accordingly, cardiac arrhythmias—especially AF or ventricular tachycardia (VT)—can limit CO and cause exercise intolerance in performance animals (see later discussion). Clinical aspects of myocardial disease are discussed later in this chapter.
Valvular and Endocardial Diseases
The cardiac chambers are lined by the endocardium, which also covers the four cardiac valves and is continuous with the endothelium of the great vessels. Normal valves govern the one-way flow of blood through the heart by preventing significant regurgitation of blood from higher to lower pressure zones. The AV inlet valves—the tricuspid and the mitral—are anchored by the collagenous chordae tendineae and papillary muscles and are supported by a valve “annulus” and the caudal atrial walls (see Figures 10-1 and 10-2).70 The mitral valve consists of two major cusps and several accessory cusps.45 The tricuspid valve is the largest valve and consists of three well-defined leaflets. Lesions of any portion of the AV valve apparatus or dilatation of the ventricle can lead to valvular insufficiency (see Box 10-2). The aortic and pulmonary valves each consist of three semilunar leaflets that close during diastole to protect the ventricles from the higher arterial blood pressure (ABP). Aortic valvular tissue in horses is not simply passive and will contract in response to a number of adrenergic and vascular agonists, such as angiotensin II and endothelin.123 The left and right main coronary arteries originate within the aortic valve sinuses (of Valsalva).
Valvular disorders in horses are common.∗ Congenital valve stenosis, dysplasia, or atresia are recognized sporadically in foals.4,111,127,155–168 Degenerative diseases of the aortic, mitral, and tricuspid valves are very common in mature horses,4,127,155–168 and endocarditis can develop on any cardiac valve† (see Box 10-2). Valvular lesions of obscure cause, including nonseptic valvulitis, have been recognized sporadically. Tricuspid and MR, of unspecified cause, is often detected in high performance animals, including Standardbred and National Hunt horses.182–185 MR caused by rupture of a chorda tendineae is recognized in both foals and mature animals.38,132,147,186 Clinical aspects of valvular heart disease are discussed later in this chapter.
Disease of the Impulse-Forming and Conduction Systems
The specialized cardiac tissues consist of the sinoatrial (SA) node, internodal pathways, AV node, bundle of His, bundle branches, fascicles, and Purkinje system (Figure 10-3). The SA node, a relatively large, crescent-shaped structure, is located subepicardially at the junction of the right auricle and cranial vena cava. Well-documented sinus node disease, although suggested,187 is rare; in contrast, vagally induced sinus arrhythmias are common.9,187–189 The equine atrial muscle mass is large and predisposes the horse to development of re-entrant rhythms and fibrillatory conduction.190 The AV node, situated in the ventral atrial septum, and the bundle of His, which continues into the bundle branches, are sites for AV block, both physiologic (vagal), and infrequently, pathologic in nature. Conduction is slow across the normal AV node.191–193 The His-Purkinje system in the ventricular septum and ventricular myocardium can act as substrates for junctional and ventricular ectopic impulses and tachycardias. Because the horse has relatively complete penetration of Purkinje fibers in the ventricles—except for a small portion of the LV free wall—the substantial equine ventricles are electrically activated in a relatively short time (≈110 msec).7
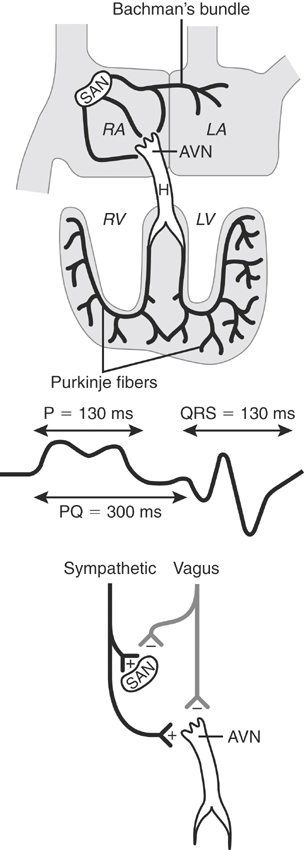
The autonomic nervous system extensively innervates the heart and influences cardiac rhythms. 4,111,134,194–199 Interplay between the sympathetic and parasympathetic branches normally controls heart rate and rhythm in response to changes in ABP.9,200 The vagus nerve innervates supraventricular tissues extensively and probably affects proximal ventricular tissues to a minor extent. Vagal influence generally depresses heart rate, AV conduction, excitability, and myocardial inotropic (contractile) state. However, because vagotonia also shortens the action potential and refractory period of atrial myocytes, high vagal activity is a predisposing factor in the development of AF.201 Innervation of the stimulatory sympathetic nervous system is extensive throughout the heart and has effects generally opposite to those of the parasympathetic system. β1-Adrenergic receptors dominate in the equine heart,202 but presumably, other autonomic subtype receptors exist, including α-adrenergic receptors and small numbers of β2-adrenoceptors.76,203 The notable increase in heart rate that attends exercise is related to increased sympathetic efferent activity and withdrawal of parasympathetic tone.9 Increases in heart rate to 220 to 240 beats/min are not uncommon with maximal exercise.204–208 The exact role of dysautonomia in the genesis of cardiac arrhythmias has not been determined; however, infusion of autonomic receptor agonists and antagonists can be associated with direct or baroreceptor-induced changes in heart rate and rhythm.209–212 Cardiac arrhythmias are discussed later in this chapter.
Vascular Diseases
There are three major subdivisions of the circulation exist: (1) systemic, (2) coronary, and (3) pulmonary. The arteries and veins consist of three layers: (1) adventitia, (2) media, and (3) intima. The overall structure and function of each layer varies with the vessel and location. Vascular receptors20,73,75,76 and anatomic lesions influence vascular resistance and blood flow. α-Adrenoceptors dominate in the systemic vasculature, and blood pressure (BP) is generally raised by vasoconstriction after stimulation of postsynaptic α-adrenergic receptors by norepinephrine, epinephrine, or infused α-adrenergic receptor agonists like phenylephrine.213,214 The presence of vasodilator β2-adrenergic receptors is clinically relevant, insofar as infused β2-agonists cause vasodilation in circulatory beds that contain high β-agonist adrenergic receptor density. Many vascular beds also dilate after the production of local vasodilator substances, such as nitric oxide, released during exercise, stress, or metabolic activity.73,75 Dopaminergic receptors, when present in vascular walls, may be stimulated, causing vasodilation, provided vasoconstricting α-adrenergic activity does not dominate. Stimulation of histamine (H1) receptors or serotonin (5-HT) receptors causes arteriolar dilatation, venular constriction, and increased capillary permeability.75 Infusion of endothelin215 or of calcium salts causes arterial vasoconstriction,216 whereas administration of calcium channel antagonists (e.g., verapamil, diltiazem) causes vasodilation of vascular smooth muscle.217
Various vascular lesions have been reported in horses (see Box 10-2). Rupture of the aorta, PA, or middle uterine artery is devastating and often lethal.∗ The aorta may also rupture into the heart, creating an aortic to cardiac fistula.220,221 Although parasitic arteritis may predispose to vascular injury, the cause of most vascular lesions, including aortic-iliac thrombosis, is unknown.222–227 Causes of vasculitis include Strongylus vulgaris infestation of the cranial mesenteric artery, infective thrombophlebitis of the jugular veins, equine viral arteritis, and suspected immune-mediated disease.106,108,228 Neoplasms can obstruct blood flow by external compression or through invasion, more often affecting the right side of the circulation. Examples include obstruction of the PA by a lung tumor and obstruction of venous return by neoplastic compression or invasion of the vena cava. Clinical features of vascular disease are discussed later in this chapter.
CLINICAL CARDIOVASCULAR PHYSIOLOGY
The clinician must appreciate elementary aspects of normal heart function to perform a clinical CV examination and understand the abnormalities associated with heart disease and CHF.229 Central to this are the electrical-mechanical correlates of Wiggers cardiac cycle.
Cardiac Cycle
The association between electrical and mechanical events of the heart first described by Wiggers has been reviewed in standard physiology textbooks (Figure 10-4).73,75–77 From a study of this cycle, it is evident that cardiac electrical activity precedes pressure and volume changes; therefore arrhythmias can exert deleterious hemodynamic effects, especially during exercise, illness, or anesthesia. Relevant aspects of this cycle are now considered.
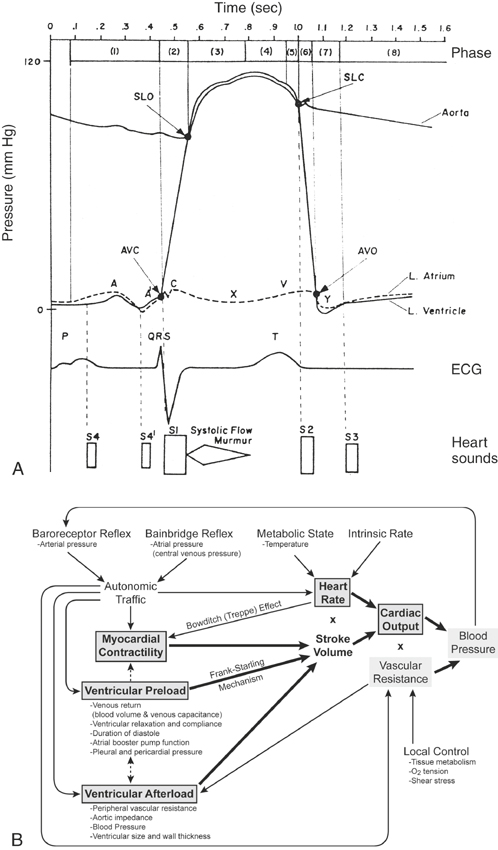
The QRS complex heralds ventricular systole. After depolarization of the ventricular myocytes, calcium enters the cell to trigger release of calcium stores in the sarcoplasmic reticulum. Increased cytosolic calcium interacts with the cardiac troponin complex on actin and myosin filaments to shorten the myofilaments and develop tension. These events are enhanced by sympathetic activity or drugs such as digoxin or dobutamine and depressed by anesthetics and drugs that impair calcium entry into cells. The abrupt increase in ventricular wall tension and chamber pressure closes the AV valves (coinciding with the vibrations of the first heart sound, S1) and increases intraventricular pressure (isovolumetric period) until the semilunar valves open.23,25 At this instant the ventricular walls move inward (Figure 10-5, B and C) and blood is ejected into the great vessel as the ventricular pressure increases to peak value and creates a similar peak ABP (see Figures 10-5, D, and 10-6). The contracting heart twists during systole, and the LV strikes the chest wall caudal to the left olecranon causing the cardiac impulse or “apex beat.” This early systolic movement, coincident with opening of the aortic valve, is a useful timing clue for cardiac auscultation and for identifying the mitral valve area for auscultation. The delay between the onset of the QRS and the opening of the semilunar valves, termed the pre-ejection period, can be measured by Doppler echocardiography and is an index of ventricular myocardial contractility such that positive inotropic drugs shorten the pre-ejection period.46,230–237 Blood is ejected into the aorta and PA with an initial velocity that generally peaks near 1 ms/sec and can be measured by Doppler echocardiography.149,230 The aortic ejection time usually exceeds 400 ms in a horse at rest, and reductions of either ejection velocity or ejection time are suggestive of reduced LV function. A functional systolic ejection murmur is often heard during ejection (see Figure 10-4, A). Such murmurs, by definition, must begin after the first sound and end before the second sound. The difference between the diastolic and systolic pressure (pulse pressure) and rate of rise of pressure contribute to a palpable arterial pulse during midsystole (see Figures 10-4, A, and 10-6). The precise timing of the pulse depends on the proximity of the palpation site relative to the heart. At the end of the ejection period, as ventricular pressures fall below those of the corresponding arteries, the semilunar valves close coincident with the high-frequency second heart sound (S2) or sounds and the incisura of the arterial pressure curves.19,23,25 The pulmonary valve may close either after or before the aortic valve.3,4,238 Asynchronous valve closure may lead to audible splitting of S2, which is normal but can be extreme in some horses with lung disease. During the ejection period the ventricular volume curve graphs a marked reduction from the end diastolic volume: this volume ejected is defined as the stroke volume. The ratio of the stroke volume to the end diastolic volume is the ejection fraction, a commonly used index of systolic heart function and correlated to the often-used shortening fraction of the M (motion)-mode echocardiogram (see Figure 10-5). Contraction of the ventricles causes the atrial pressures to decline leading to the x descent of the atrial pressure curve and a brief systolic collapse of the jugular vein. Subsequent to atrial filling during ventricular systole, a positive pressure wave, the v wave, occurs in the atrial and venous pressure curves. Severe TR accentuates this wave and may lead to pathologic systolic pulsations extending up the jugular furrow. Finally, a decline in ventricular pressure (isovolumetric relaxation) occurs, related to off-loading of calcium from the troponin apparatus and resequestration into the sarcoplasmic reticulum. This active ventricular relaxation is associated initially with closure of the semilunar valves and eventually by opening of the AV valves. Relaxation in healthy hearts is enhanced by sympathetic activity. Conversely, myocardial ischemia can impair active relaxation, and it is likely that subendocardial ischemia combined with reduced diastolic filling time contribute to the marked elevations in LA pressures observed during galloping or other high-intensity exercise.239,240
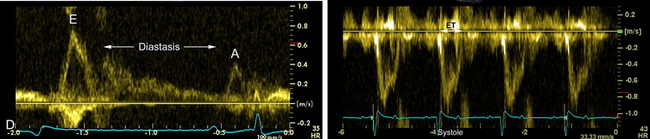
Ventricular filling commences just as the AV valves open. As shown in Figure 10-5, D, ventricular diastole can be subdivided into three general phases: (1) rapid ventricular filling, (2) diastasis, and (3) atrial contraction.73,75,76 These phases are readily observed using pulsed-wave Doppler echocardiography or Doppler tissue imaging.241 Once the ventricles have relaxed and the atrial pressure exceeds the corresponding ventricular pressure, the AV valves open. At that instant, rapid filling ensues with a peak velocity of about 0.5 to 1 m/sec, but varying directly with the heart rate.149 The ventricular pressures increase only slightly during this phase, whereas the ventricular volume curves change dramatically from the venous return. Rapid filling may be associated with a functional protodiastolic murmur, which is concluded by the third heart sound (S3), the low-frequency vibrations occurring near the termination of rapid ventricular filling. The loss of atrial volume and corresponding decline in the atrial pressure (the y descent) is reflected in the jugular furrow as the vein collapses. After rapid filling, a period of greatly reduced low-velocity–filling diastasis ensues. This period may last for seconds during vagal arrhythmias such as sinus bradycardia, pronounced sinus arrhythmia, or second-degree AV block. With markedly exaggerated pauses, the jugular vein may begin to fill prominently. The last phase of diastole is the contribution to ventricular filling caused by the atrial contraction. A functional presystolic murmur has been associated with this period between the fourth and first heart sounds.
During the cardiac cycle the atrium functions as a reservoir for blood (ventricular systole), a conduit for venous return (early to mid-diastole), and as a pressure pump (atrial systole). Mechanical atrial function of the LA can be studied using two-dimensional (2D) echocardiography and advanced Doppler echocardiographic methods. Impaired electrical and mechanical function of the atria may predispose to recurrent atrial arrhythmias such as AF.190,242–245
ASSESSMENT OF VENTRICULAR FUNCTION
The ability of the ventricles to eject blood depends on both systolic and diastolic ventricular function, as well as heart rate and rhythm (Box 10-3). The most commonly used measurements of overall ventricular performance and circulatory function are invasively or noninvasively determined ABP, rate of ventricular pressure change (dp/dt), CO, stroke volume, ejection fraction, LV shortening fraction, systolic time intervals, central venous pressure, PA or wedge pressures, and arteriovenous oxygen difference (A-V DO2).∗ CO, the amount of blood pumped by the left (or right) ventricle in 1 minute (L/min), is the product of ventricular stroke volume (ml/beat) multiplied by the heart rate (beats/min; see Figure 10-4, B). Cardiac index refers to the CO divided by (indexed to) the body surface area (or body mass). CO coupled to systemic vascular resistance determines the mean ABP: an increase in either variable raises mean arterial pressure. Values for CO vary widely with the size and activity of the horse and are often influenced by drug therapy or anesthesia.210,260–275 A noninvasive estimate of CO can also be obtained using Doppler techniques.233,234,257,276–285
BOX 10-3 DETERMINANTS OF CARDIAC FUNCTION
Systolic Function: Determinants of Ventricular Stroke Volume
Ventricular stroke volume depends on myocardial contractility and loading conditions (preload and afterload; see Figure 10-4, D).24,26 Although traditionally considered independent determinants of myocardial function, these variables are all interconnected and influence force, velocity, and duration of ventricular contraction.76 Ultimately, the availability of calcium to the sarcomere is modulated by the inotropic state, the initial myocardial stretch (preload), and the tension that must be generated to eject blood into the vascular system (afterload).
Myocardial contractility is increased by catecholamines, calcium, digitalis glycosides, and phosphodiesterase inhibitors.∗ Contractility is difficult to measure in the clinical setting but can be estimated noninvasively by observing directional changes in load-dependent pre-ejection or ejection phase indices of ventricular function. These include shortening and ejection fractions by M-mode and 2D echocardiography; pre-ejection period, ejection time, flow acceleration, velocity time integral of aortic or pulmonic ejection, and peak myocardial velocity by Doppler echocardiography; and myocardial deformation or strain by computerized analysis of 2D echocardiograms or tissue Doppler studies (see Figure 10-5).49,50,51,237,303–306 Measured variables will be influenced by physiologic state, altered mildly by day-to-day variation307,308 or sedatives,231 and affected markedly by general anesthesia.
Ventricular fiber length or preload is a positive determinant of ventricular systolic function that depends on venous return and ventricular size and distensibility. The normal ventricle is highly preload dependent, such that increases in preload increase stroke volume. Dehydration, venous pooling, loss of atrial contribution to filling (AF), and recumbency all reduce ventricular filling and decrease stroke volume. When hypotension develops consequent to decreased ventricular filling, intravenous (IV) crystalloid or colloid is often administered initially to increase venous pressure and ventricular preload. Increased preload also can be observed in horses with heart disease as a consequence of impaired pump function, cardiac lesions, or fluid retention. Moderate to severe valvular insufficiency increases ventricular filling pressures and preload.38,105,309–311 The increased ventricular diastolic dimension serves as a compensatory mechanism that maintains forward stroke volume in the setting of a failing ventricle or regurgitant heart valve.
Ventricular preload can be estimated by determining ventricular end diastolic volume or size, as measured by echocardiography, or by measuring venous filling pressures.310 The measurement of venous filling pressures (central venous pressure, pulmonary diastolic or pulmonary capillary wedge pressure)310–314 provides an accurate gauge of preload provided that heart rate and ventricular compliance (distensibility) are normal and ventilation is relatively stable. Myocardial ischemia, which impairs myocardial relaxation, and pericardial diseases, which constrict the ventricles, both reduce ventricular compliance; in such cases, the venous filling pressures may not accurately reflect chronic changes in ventricular preload.
Ventricular afterload is represented by the ventricular wall tension that must be developed to eject blood and relates to forces impeding this ejection. Peak wall tension occurs immediately before aortic valve opening. Vascular impedance during ejection also relates to the elastic, resistive, and dynamic properties of the connected great vessel and vascular tree. Increases in ventricular chamber size, arterial stiffness, or resistance, as well as marked increases in hematocrit, increase the impedance to ventricular ejection and reduce stroke volume. Afterload is difficult to measure clinically, and although BP is not identical to afterload, it may be used to estimate directional changes in afterload. LV failure, alveolar hypoxia-induced pulmonary vasoconstriction, atelectasis, and PA or venoocclusive diseases are important causes of increased RV afterload. Arterial vasodilators such as hydralazine and angiotensin converting enzyme (ACE) inhibitors decrease afterload.268,315
Ventricular synergy refers to the normal method of ventricular activation and contraction. Normal electrical activation causes a burst of activation of great mechanical advantage.20 Cardiac arrhythmias, especially ventricular rhythm disturbances, can cause dyssynergy (dyssynchrony), with a resultant decrease in stroke volume. Coronary occlusions leading to ischemic myocardial necrosis also cause dyssynergy but are considered relatively rare.57,108,316
Ventricular diastolic function determines ventricular filling and preload.73,75,76,259 Factors that affect diastolic function are indicated in Box 10-3. When diastolic function is abnormal, often greater heart rate and higher venous pressure dependencies occur for maintenance of CO. A well-recognized cause of diastolic dysfunction is constriction or compression of the heart caused by pericardial disease. Marked ventricular chamber dilatation or hypertrophy also decreases ventricular compliance and requires higher ventricular distending pressures for filling. LV diastolic dysfunction as a consequence of severe RV dilatation or hypertrophy can be explained by bulging of the ventricular septum into the LV, which impedes left-sided filling. This influence of ventricular interdependence is observed clinically with chronic pericardial disease and severe pulmonary hypertension. Ventricular diastolic function also is affected by arrhythmias. Persistent tachycardia shortens diastole, cardiac filling time, and coronary perfusion. With AF, the atrial contribution to filling is lost. Junctional and ventricular arrhythmias lead to AV dissociation preventing normal AV sequencing and can also create marked dyssynchrony of ventricular contraction.
Imbalance between myocardial oxygen demand and delivery can reduce both ventricular systolic and diastolic function and may affect cardiac rhythm as well. This relationship is also relevant when there is airway obstruction or bronchopulmonary disease, which can reduce arterial oxygenation.317 Myocardial oxygen demand is augmented by increasing myocardial inotropic state, heart rate, and ventricular wall tension (related to preload and afterload).76 Oxygen delivery depends on coronary anatomy and vasomotion (degree of vessel constriction), diastolic ABP, diastolic (coronary perfusion) time, and metabolic activity of the myocardium.10,75,318–325 Normal coronary flow is highest to the LV myocardium in the ventricular septum and LV wall.325 The immediate subendocardial layer of myocardium is probably most vulnerable to ischemic injury,9,318
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


