(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
It is time now to address the most important question as to why T2D is largely incurable or irreversible. There are three possible reasons for the apparent irreversibility. It is possible that the pathophysiological changes are irreversible by nature. Alternatively, we know what needs to be done in order to cure diabetes, but we do not have the right drugs or the technology to do it. The third possibility is that we do not know what the real cause is, and therefore, we are trying to hit a target that has nothing much to do with the disease. I am going to argue below that the third one is true. All the antidiabetic drugs currently in use and the new candidates in various stages of R&D are hitting at the wrong target and therefore are bound to be ineffective in principle. The problem does not lie in not having the technology to mend what is wrong. The problem lies in identifying what is really wrong. If insulin resistance is central to the pathophysiology of T2D, then efficient insulin-sensitizing drugs are available. But insulin-sensitizing drugs do not cure diabetes, and therefore, we need to suspect whether insulin resistance is really at the root of the pathophysiology of diabetes. If we knew that insulin resistance was the key to T2D but did not have any drug to reduce insulin resistance, that condition would have been comparable to beating around the bush. But the emerging picture is not this. We are unable to cure diabetes not because we do not have insulin-sensitizing drugs. We have them and we also know that exercises are insulin sensitizing. But still medicine has failed to prevent or cure diabetes. This situation is worse than beating around the bush, and I will call it beating around the wrong bush. It is not very difficult to see why I say this if we look at some of the recent evidence accumulating on the broader horizon of insulin action and insulin sensitivity across the animal world.
It is time now to address the most important question as to why T2D is largely incurable or irreversible. There are three possible reasons for the apparent irreversibility. It is possible that the pathophysiological changes are irreversible by nature. Alternatively, we know what needs to be done in order to cure diabetes, but we do not have the right drugs or the technology to do it. The third possibility is that we do not know what the real cause is, and therefore, we are trying to hit a target that has nothing much to do with the disease. I am going to argue below that the third one is true. All the antidiabetic drugs currently in use and the new candidates in various stages of R&D are hitting at the wrong target and therefore are bound to be ineffective in principle. The problem does not lie in not having the technology to mend what is wrong. The problem lies in identifying what is really wrong. If insulin resistance is central to the pathophysiology of T2D, then efficient insulin-sensitizing drugs are available. But insulin-sensitizing drugs do not cure diabetes, and therefore, we need to suspect whether insulin resistance is really at the root of the pathophysiology of diabetes. If we knew that insulin resistance was the key to T2D but did not have any drug to reduce insulin resistance, that condition would have been comparable to beating around the bush. But the emerging picture is not this. We are unable to cure diabetes not because we do not have insulin-sensitizing drugs. We have them and we also know that exercises are insulin sensitizing. But still medicine has failed to prevent or cure diabetes. This situation is worse than beating around the bush, and I will call it beating around the wrong bush. It is not very difficult to see why I say this if we look at some of the recent evidence accumulating on the broader horizon of insulin action and insulin sensitivity across the animal world.
Wrong Bush 1: Insulin Resistance
The protein insulin and its equivalents are widespread in the animal world including vertebrates and invertebrates. Since impairment of insulin signaling or insulin resistance is believed to be central to a cluster of deadly diseases in humans, it would be useful to look at what happens when there is impairment of insulin signaling in other animals. And here lies a very fundamental paradox. In widely different taxa of animals, including both invertebrate and vertebrates, impairment of insulin signaling has no adverse effects on health; on the contrary, it increases life span. This is one very fundamental and perplexing fact reviewed very well by Kaletsky et al. [1].
Over 20 years ago, it was discovered that mutations in daf-2 and age-1 double the life span of a nematode worm model Caenorhabditis elegans [2, 3]. Subsequent cloning of these genes uncovered the importance of the insulin/insulin-like growth factor-1 (IGF-1) signaling pathway in aging regulation. daf-2 encodes the only insulin/IGF-1 receptor expressed in worms, and age-1 is the catalytic subunit of the downstream phosphoinositide 3-kinase (PI3K) [4, 5]. Mutations in these genes interfere with the normal insulin signaling pathway and therefore are equivalents to insulin resistance in humans. These “insulin-resistant” worms have a substantially longer life span. In addition to the overall life span extension, worms bearing either of these mutations, i.e., the insulin-resistant worms, are highly resistant to oxidative stress [6], hypoxia [7], heat [8], heavy metals [9], and bacterial pathogens [10].
The insulin/insulin-like growth factor signaling (IIS) pathway also acts as a food and stress sensor during development. When food is abundant, worms develop rapidly and uninterrupted through four larval stages to reach adulthood. If worms develop in food-limited or overcrowded conditions, they enter an alternative long-lived larval state called dauer in which reproductive maturity is delayed but resistance to environmental challenges increases. Favorable nutritional conditions promote exit from dauer which is again mediated by changes in insulin signaling pathway and development to reproductive adults [11]. The daf-2 mutants are dauer phenotype by default [12]. Both the long-lived and dauer phenotypes of daf-2 worms are dependent on the downstream forkhead transcription factor DAF-16 [2, 12, 13]. Briefly speaking, signaling through DAF-2 activates PI3K, which leads to the phosphorylation of DAF-16 and its inactivation by nuclear exclusion [14]. In the absence of IIS or in daf-2 or age-1 mutants, DAF-16 enters the nucleus and enacts a transcriptional program that doubles worm life span [10, 15–17]. Loss of daf-2 or age-1 also slows the age-related declines; thus, the insulin signaling pathway regulates longevity through its modulation of processes and pathways leading to aging.
This is not a worm story alone. Insulin signaling is an evolutionarily conserved pathway that regulates life span across many species. Drosophila melanogaster has a single insulin-like receptor (dInR) that, when mutated, extends life span in a manner that is dependent on its daf-16 homolog dFOXO or simply foxo [18]. Overexpression of dFOXO in the fat body [19] or mutation of the insulin receptor substrate protein chico extends life span in the fly [20, 21]. Mammals encode several daf-2 homologs including IGF-1 receptor (IGF-1R) and insulin receptor (IR)-A [22]. Despite these differences in insulin receptor expression, the functional consequences are similar, as reduced insulin/IGF signaling extends life span in multiple mammalian species [23, 24]. Longevity in dogs is inversely proportional to body size [25]. Interestingly, a single nucleotide polymorphism in IGF-1 is associated with small size and long life, suggesting that IIS influences aging in dogs [26]. Heterozygous IGF-1R knockout mice are long lived [27], and adipose tissue-specific insulin receptor knockout (FIRKO) mice also exhibit increases in life span [28, 29].
In mammals, there are some counterexamples where low insulin production and insulin sensitivity are associated with long life. Naked mole rats, known for their exceptionally long life spans, are insulin sensitive [30]. Recent studies suggest that IIS and aging are linked processes in mammals and specifically humans as well. But there appears to be a contradiction in the direction of the association. It is well known that insulin resistance is associated with a large cluster of disorders, many of which are fatal and therefore should reduce life span. Insulin sensitivity is a common feature of centenarians. Mutations in a human daf-16 homolog, FOXO3a, are linked to increased longevity [31–33]. Long-lived men who are homozygous for the FOXO3a GG genotype also display greater insulin sensitivity [31]. Therefore it may appear that insulin sensitivity rather than insulin resistance is the key to longevity in humans. We may conclude that sometime in evolution, the relationship might have turned upside down.
But unfortunately this inference is not so straightforward. Other pieces of evidence point to the possibility that similar to worms and flies, insulin resistance increases life span in humans too. IGF-1R mutations are highly represented in populations of centenarians [34] indicating impaired IIS associated with long life. Of particular importance is the gene called Klotho known to be a longevity gene in mammals [35]. Klotho, the gene coding for a transmembrane enzyme, was discovered in 1997. Overexpression of this gene increases life span, and mutants with impaired expression age prematurely. It was therefore named after one of the three Greek Moirae or fate called Clotho that weaves the thread of life. It was soon discovered that Klotho increases life span by inducing insulin resistance. Klotho induces adipocyte differentiation [36], lipogenesis [37], and insulin resistance [38, 39]. Contrasting with this stands out the fact that Klotho increases life span substantially. There are two different paradoxes here. First is that although almost every model of longevity extension involves insulin signaling, the direction appears confusing. In some examples longevity is associated with increased insulin sensitivity and in other models insulin resistance. The emerging solution to the paradox is that it is reduction in insulin signaling at a subcellular level that increases life span. This can be achieved either by lower levels of insulin or by reducing the sensitivity to insulin. If this turns out to be true, no real paradox is left since in long-lived phenotypes that are insulin sensitive such as naked mole rat, insulin production is indeed very low.
The concept of insulin resistance increasing life span is actually theoretically very sound at both proximate and ultimate levels. We have argued before that insulin resistance is a mechanism of modifying reproductive strategies to adapt to the demography and environment. Typical r reproductive strategy is insulin sensitive, and K reproductive strategy is insulin resistant. As a broad generalization across species, K-selected species are generally long lived as compared to r-selected species. We do not know whether this would be true for reproductive strategies within a species as well. But theoretically it should and therefore insulin-resistant individuals should be long lived as expected by life history theories. It is interesting to note that although the Klotho protein is essential for fertility, as demonstrated by infertility of Klotho knockouts [40], Klotho overexpression actually reduces fecundity [41]. Also note that placenta produces Klotho protein [42] which by inducing insulin resistance can draw more glucose through the placenta. Therefore Klotho appears to be the ideal K reproduction mechanism, one that increases longevity, reduces fecundity, and increases transplacental investment in the fetus through insulin resistance.
Insulin is a growth hormone during developmental period [43, 44]. Insulin has modulating effects on reproductive strategies [45–48], and insulin is an important modulator of life span as seen above. All three are important parameters of life history evolution. This means that insulin is the chief mediator of life history strategies. Being a fine-tuner of aggressive behavior, it also serves the function of selecting behavioral strategies for an individual. Aggression cannot be separated from life history strategies. Presence of predators is well known to shape life history strategies. Having aggressive competitors would play a role similar to predators. Since both predation and aggression increase the probability of death, life histories will tend more towards r and away from K. Therefore, insulin’s role in aggression cannot be separated from its role in life history strategies. This appears to be the main role of insulin conserved across simple to complex animal taxa. Insulin’s role in glucose homeostasis is only a small part of this bigger role. Just because insulin’s role in glucose homeostasis was discovered earlier, medicine believed that that was the primary function of insulin. An evolutionary perspective shows that this is a grossly incomplete and biased view. Understanding insulin’s function on a broader canvas can certainly deepen our understanding about insulin and thereby give us greater insights into diabetes too. Without understanding life history strategies and life sustenance strategies, it is impossible to understand insulin. Traditional medicine has been doing precisely this mistake so far and that must have contributed to the overall failure to prevent, control, or cure diabetes.
This leads us to the other paradox. How is insulin resistance associated with increased life span when it leads to so many deadly disorders? Here, once again, Klotho shows the way out. The paradox gets resolved when we look at the other metabolic and endocrine effects of Klotho. Klotho has anti-inflammatory effect [49], proangiogenic action [50], proendothelial function [51], and antioxidant activity [52]. We have seen earlier and will elaborate once again below that inflammatory changes, endothelial dysfunction, angiogenesis dysfunction, and oxidative damage are the main pathophysiological mechanisms of T2D. The prevalent perception is that insulin resistance or hyperglycemia is the driver of these pathological effects. Since in diabetes insulin resistance and/or hyperglycemia is invariably coupled with its pathophysiological mechanisms, the association has been assumed to be mandatory. A fundamental conceptual revision that Klotho has necessitated is that the association is not mandatory. Here is a mechanism in the form of Klotho protein that can decouple obesity and insulin resistance from the true pathological mechanisms of systemic inflammation, endothelial and angiogenesis dysfunction, and oxidative damages. Having thus decoupled, insulin resistance appears to increase longevity.
The inference that one can draw from Klotho research is that insulin resistance is not sufficient to drive the pathophysiological mechanisms of T2D. The real pathological drivers are different. In fact insulin resistance may be good for health if there is a way to decouple it from the true pathological drivers. Klotho is certainly an effective decoupler, but perhaps there can be other mechanisms too. Once we know that such a decoupling is possible in principle, researchers can specifically look for other mechanisms. The decoupling also means that if the pathophysiological mechanisms are not targeted, targeting insulin resistance will be ineffective as a strategy for the treatment of T2D. It may appear to normalize blood sugar temporarily but is most unlikely to cure diabetes because insulin resistance is not central to the pathophysiology of diabetes at all. Insulin resistance is the target for the mainstream pharmacological R&D in search of a wonder drug against T2D. A generation of insulin-sensitizing drugs is already in the market and being used commonly, but T2D remains an “incurable” disorder. This is because all our efforts are not only beating around the bush but beating around the wrong bush. Although insulin resistance is central to the behavioral transition from soldier to diplomat, it is not likely to be the root cause of T2D and related pathology, and therefore, targeting insulin resistance will never cure diabetes.
Wrong Bush 2: Hyperglycemia
The orthodox paradigm has revolved only around glucose and insulin and as a result may have completely misled medicine for decades together. Plasma glucose is an important parameter in diagnosis, and it will remain so for some more time. However, doubts can be raised about its value as an indicator of the effect of treatment and a predictor of pathological complications. First of all one should differentiate between hyperglycemia and diabetes. Hyperglycemia is only one of the symptoms of diabetes. The current treatment regime has the sole aim of normalizing plasma glucose, and therefore, it does not really treat diabetes. All that it can control is hyperglycemia. There is a strong prevalent belief that controlling sugar levels are sufficient to prevent all types of diabetic complications. Evidence is not completely in favor, and therefore, this postulate needs to be critically reexamined as we have discussed earlier (Chap. 3).
The paradigm that hyperglycemia is the root of all pathophysiology of T2D is an old one and has been there with us for several decades. Interestingly, in spite of decades of research, the mechanisms by which glucose can bring about wound-healing problems, peripheral neuropathy, endothelial dysfunction, and erectile dysfunction have never been clearly and completely worked out. All that we have are some incompletely worked out mechanisms, some other hypotheses that were never put to test, and some prevalent beliefs that are not even worth calling hypotheses. As a good example of the latter, it was believed that increased sugar encourages bacterial growth, and therefore, wound healing is difficult and infections are more common in diabetes. However, honey has been traditionally used as a wound-healing agent, and a number of careful studies have reproducibly demonstrated the wound-healing properties of honey [53, 54]. Here is an example of high sugar concentration not interfering in wound healing. Why should sugar interfere in wound healing only in diabetes? But this is a story that is popularly believed in the layman’s circle, and even some practicing physicians seem to subscribe to it though fortunately not many researchers.
Glucose is proposed to cause a series of pathological changes through proposed mechanisms that we saw in Chap. 2. This is the current mainstream thinking. But the Klotho story suggests something else. Impaired Klotho pathway can give rise to a phenotype which is lean, insulin sensitive, and normoglycemic but shows rapid age-related degenerative changes including oxidative damage, chronic systemic inflammation, and vascular problems similar to those in diabetes. Since the pathological components of T2D such as inflammatory changes, oxidative excess, and endothelial and angiogenesis dysfunctions can be demonstrably decoupled from glucose homeostasis mechanisms, they are likely to have origins independent of glucose levels. This could imply that these pathological processes could also be ameliorated without worrying about glycemic control.
In order to elaborate on the possibility of glucose-independent origins of diabetic pathology, I will put together several pieces of evidence for the same set of pathological changes being caused by behavior-driven pathways. These alternative pathways work independent of sugar but are not incompatible with the sugar-driven pathways. We will see the alternative pathways first and then think of any convergence or contradiction between the behavior-driven and glucose-driven pathways.
This new landscape of behavior-driven pathology is more complex than the orthodox picture but much more realistic and evidence based. Although there are still some loose ends left where more research is needed, it is much more detailed than the orthodox one. Here the root of all pathophysiology lies in brain and behavior, not blood sugar. The brain and endocrine systems of a soldier-deficient or supernormal diplomat phenotype send independent signals that bring about simultaneous changes in (1) sex and aggression signals; (2) insulin secretion; (3) fat accumulation; (4) immune redistribution; (5) disinvestment from wound healing and angiogenesis mechanisms; (6) deficiencies of EGF, NGF, BDNF, and many other growth factors; and (7) deficiency of erythropoietin (Fig. 13.1). We have already seen the origin of these alterations at both ultimate and proximate levels, but a brief reiteration would be appropriate. Long-term absence of aggressive encounters reduces levels of aggression signals including testosterone. Higher insulin levels are needed by the brain to provide for the greater demand for cognitive functions. This is mediated by the parasympathetic stimulation of pancreas. High insulin activity induces fat accumulation. The reduced frequency as well as reduced anticipation of injuries leads to disinvestment from wound healing and angiogenesis mechanisms as well as accumulating deficiency of erythropoietin and growth factors. Reduced injuries along with increased levels of adipokines mediate redistribution of macrophages. The seven primary changes are partially independent but have interactions and mechanisms reinforcing each other. For example, testosterone deficiency facilitates adiposity and arrests EGF synthesis. Adipose tissue facilitates altered distribution of macrophages and so on. However I will still call all seven processes primary since they can also originate independently in the absence of others.
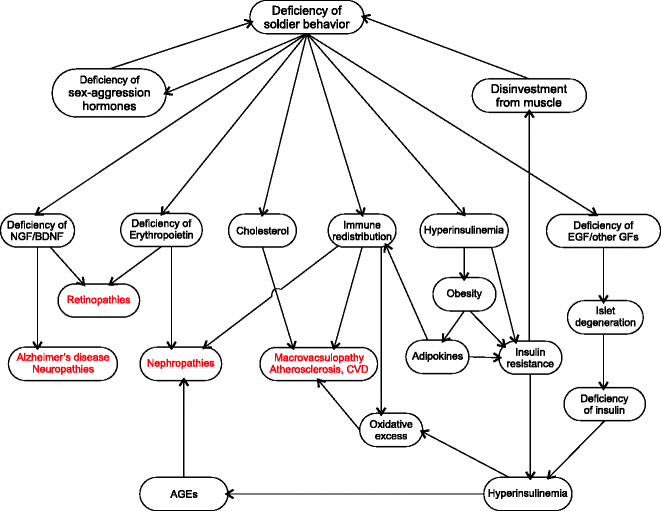

Fig. 13.1
Pathophysiology of diabetic complications: The new paradigm suggests a different route to diabetic complications that incorporates the effects of hyperglycemia but does not assume hyperglycemia to be the root cause of all complications. The alternative pathways account for many of the horse and cart paradoxes and are substantially supported by evidence. This line of thinking can potentially bring about major shifts in diabetes management and treatment (see text for detailed explanation)
These half a dozen primary processes stemming independently from a common neurobehavioral origin trigger a chain of processes downstream (Fig. 13.1). Downstream to hyperinsulinemia, signals from adipose tissue, pancreas, and perhaps brain induce compensatory insulin resistance. Disinvestment from angiogenesis reduces capillary density which also reduces glucose uptake by muscle and other tissues contributing to insulin resistance. Immune redistribution leads to vascular and systemic inflammatory changes as well as oxidative excesses. Deficiency of EGF and other growth factors leads to progressive β cell loss [55, 56]. Reduced insulin levels lead to disinvestment from muscle on the one hand and increased plasma glucose on the other. As a result of disinvestment from muscle, soldier behavior becomes increasingly impossible, and this reinforces diplomat behavior completing the circle and thereby stabilizing the state.
The emerging alternative picture can explain each of the diabetic complications with a sound logic and substantial support from available evidence, although these hypotheses still remain to be tested directly.
1.
Beta cell degeneration: The new paradigm suggests three independent causes for β cell degeneration. First is behavior-induced deficiency of growth factors needed for β cell regeneration. The effect of aggression on EGF secretion is well known [57]. The role of EGF in β cell growth has also been demonstrated [55, 56]; therefore, it is likely that EGF deficiency accumulating due to chronic aggression suppression is the main cause of progressive β cell degeneration as argued earlier. A little later, I will elaborate more on other growth factors required for β cell synthesis and how behavior can alter the supply of these growth factors too. The other possible cause is oxidative damage. Beta cells are unusually sensitive to oxidative damage [58–60], and Rashidi et al. think that the higher sensitivity of β cells to oxidative damage is an evolutionary adaptation [59, 60]. When the altered distribution and chronic activation of macrophages lead to oxidative excess, β cells are among the first to suffer. With altered aggression there is deficiency of sex hormones too. Testosterone and estrogens have antioxidant action, and estrogens have been shown to preserve islet function [61]. A third possible reason is that the reduced glut-1 levels chronically suppress insulin production following which β cells experience disuse atrophy. The three prospective causes are mutually compatible and might have differential contributions to the islet pathology.
2.
Macrovascular pathologies: The vascular pathologies are partially the result of immune redistribution where monocyte–macrophages accumulate within blood vessels as a result of loss of aggression and injury proneness (see Chap. 8 for details). The accumulation of macrophages in vessel walls is further aggravated by cholesterol which is also a marker of nonaggressive behavior. The other component in vascular pathology is NO deficiency which is also likely to be behavior induced. Endothelial NO synthase-deficient mice lose aggression [62] indicating that eNOS is involved somewhere in the aggression pathway itself. Since aggression involves violent muscle activity and also anticipates injuries, a fine control on vasoconstriction is needed to ensure sufficient blood flow to muscle and simultaneously minimize bleeding in case of an injury. It is crucial therefore that aggression should not be initiated unless the NO function is normal (see Chap. 7 for details). Such a behavioral checkpoint would be crucial for initiating aggression. It is likely therefore that loss of aggression may degenerate NO regulation. Sex hormones also have effects on NO-related mechanism. Estradiol induces calcium-dependent eNOS [61, 63, 64]. Testosterone has NO dependent as well as independent vasodilatory effects [65–74]. Therefore deficiency of sex and aggression signals could be a significant contributor to vascular pathology. Through the possible synergy of these mechanisms, endothelial dysfunction can precipitate independent of blood glucose. In the course of insulin resistance syndrome, signs of endothelial dysfunction appear much before diabetic hyperglycemia. Some have suggested that endothelial dysfunction is antecedent and causal rather than consequential [75]. However these processes are not incompatible with the traditional notion of hyperglycemia-induced oxidative excess at the endothelial level. Hyperglycemia may further hasten and worsen the process. But hyperglycemia does not appear to be essential for macrovascular pathology since the latter is not specific to diabetes, and tight glycemic control does not appear to be helpful in restoring endothelial function and reversing macrovascular pathology in all trials (see Chap. 3).
3.
Degenerative changes in central and peripheral nervous system: In the alternative interpretation, diabetic neuropathy is a direct result of deficiency of NGF and other neurotrophins which arises from aggression deficiency. Aggression stimulates both EGF and NGF secretion by the salivary glands [76]. EGF and NGF together stimulate BDNF expression [77], and BDNF has strong neuroprotective effects [78]. Testosterone has also been shown to have protective action against oxidative stress-induced nerve damages [79, 80].
Alzheimer’s disease appears to have multiple components. Traditionally amyloid deposits have been blamed for AD [81, 82]. The mechanism of deposition as well as its exact role in the etiology of T2D is debated. But there are alternative possibilities compatible with the new interpretation. Glut-1 downregulation is associated with AD [83]. Insulin deficiency in the brain following β cell loss or sympathetic suppression of insulin production is likely to play a causative role [84, 85], and insulin administration has ameliorating effects in AD [85–90]. NGF and BDNF deficiencies are also likely to be involved [91–99]. Along with BDNF, TrkB expression is also downregulated in AD [98]. Physical activity and aggression have been shown to be important stimulators of TrkB expression [100, 101] suggesting that deficiency of physical activity and aggression may play a direct role in AD. In an evolutionary perspective, deficiency of such hunter-gatherer behaviors is suggested to be important in the etiology of AD [102]. Being open to these alternative pathways may speed up the process of understanding AD.
4.
Nephropathy: As compared to macrovascular pathologies, microvascular pathologies appear to be more tightly linked with hyperglycemia. Unlike atherosclerosis and CVD which are not specific to diabetes, microvascular pathologies are rarely seen in the absence of diabetes. In contrast with macrovascular pathologies, the effect of tight glycemic control on nephropathy is more consistently positive across studies. However sugar alone does not seem to account for all the pathology since tight glycemic control does not completely eliminate nephropathy. The risk is reduced by 10–30 % in different studies (see Chap. 3). This suggests that apart from glucose, there could be other pathophysiological mechanisms active in diabetic nephropathy. The glucocentric view does not explain certain patterns. Erythropoietin deficiency has been shown to precede nephropathy [103], and this remains unaccounted for in the glucocentric pathophysiological pathway.
We can have a look then at the alternative picture. It is agreed that abnormal angiogenesis marks histological changes in diabetic nephropathy [104, 105]. Macrophage accumulation has been shown to be an early event in DN. Erythropoietin deficiency anemia also commonly precedes DN. Based on this we can synthesize the possible process. Erythropoietin deficiency anemia creates pockets of hypoxia which stimulate angiogenesis. Macrophages are important mediators of angiogenesis; therefore, wherever there is greater congregation of macrophages, excessive angiogenesis is more likely. Since in T2D macrophages accumulate in highly vascular tissues such as kidney and lungs, they are more prone to microangiopathy. Microangiopathy of lungs is common in diabetes for similar reasons, although fortunately serious complications are rare [106, 107]. A compelling evidence for early and fundamental involvement of macrophage accumulation in kidneys is the finding that MCP-1 knockouts or macrophage scavenger receptor A-deficient mice are resistant to diabetic nephropathy [108, 109]. Along with adipose tissue, kidneys are also involved in mediating redistribution of macrophages. Kidneys produce chemokines that alter innate immune cell dynamics in the insulin-resistant state. In this process excess macrophages accumulate in the kidneys. This excessive macrophage accumulation triggers a supernormal angiogenesis response on being stimulated by erythropoietin deficiency-mediated pocketed hypoxia. However aggressive behavior strengthens angiogenetic pathways, and in deficiency of aggression, there can be partial disinvestment from angiogenesis mechanisms. Therefore although angiogenesis is stimulated in pockets, the angiogenetic mechanisms are dysfunctional. This leads to excessive but defective angiogenesis. This abnormal angiogenesis underlies diabetic nephropathy.
5.
Retinopathy: Diabetic retinopathy may also arise from growth factor deficiency combined with erythropoietin deficiency as follows. The role of EGF and NGF in preventing retinal degeneration is well demonstrated. In tissues where there is no direct blood supply, oxygen needs to be transported by cell-to-cell diffusion. Subacute degenerative changes in the retina may impair cell-to-cell diffusion of oxygen creating pockets of hypoxia. Erythropoietin deficiency anemia can increase the chances of localized hypoxia. The resulting hypoxia stimulates abnormal angiogenesis which is the main process underlying diabetic retinopathy [110, 111]. In retinopathy it is hard to visualize how, if at all, sugar plays a role.
6.
Impaired wound healing: Wound healing is a complex process and involves a large number of signaling pathways. Many of them may be affected in diabetes. The factors that are known to be affected are NGF and EGF availability, reduced macrophage density in the subcutaneous tissues [112], and decreased angiogenesis response [113]. All three appear to be independent of sugar levels.
All the glucose-independent pathophysiological pathways proposed above are evidence based in the sense that every component of these processes has been demonstrated in the cited references. So far this work was piecemeal, and I have tried to put the pieces together to construct a coherent picture. Admittedly evidence for every step is not necessarily evidence for a ladder. But this is where more research is needed.
Agreeably there is evidence for hyperglycemia-driven pathology too. These pathways mainly involve alterations in NO expression, oxidative damages, and AGE-RAGE pathways (see Chap. 2 for details). If two alternative pictures explain the same pathology, we need some ways to resolve between the two. For example, oxidative state is implicated in both but the proposed origin of free radicals is different. This can possibly help us in resolving between the two hypotheses. By the glucose-driven view, oxidative state originates from glucose loading of cell respiratory mechanisms. Glucose loading is unlikely to happen in insulin-resistant cells but can happen in insulin-independent cells. On the other hand macrophage-driven oxidative stress will be distributed according to the distribution of macrophages. This suggests a differential testable prediction. If glucocentric hypothesis is true, insulin-independent tissues such as brain and endothelium should generate almost all the free radicals. If the macrophage hypothesis is true, origin of free radicals should be in tissues where macrophages accumulate. Macrophages accumulate in adipose tissue and vascular endothelium. Since vascular endothelium is common for both, it does not help in resolving. Adipose tissue-related predictions are in the opposite directions and therefore would be useful. Since adipose tissue is insulin dependent, it will not experience intracellular glucose excess and thereby a respiratory burst leading to free radical generation if the glucose hypothesis is correct. However since adipose tissue attracts macrophages in large numbers, it will have macrophage-generated free radicals if the alternative hypothesis is correct. If adipose tissues in T2D generate little free radicals, glucocentric hypothesis would be supported, and if adipose tissue generates larger amounts of free radicals, the macrophage hypothesis gets greater support. As we know today adipose tissue does generate substantial oxidative excess from an early stage and arresting oxidative stress in adipose tissue prevents shift of the adipokine imbalance towards proinflammatory state [114, 115]. Therefore oxidative stress in adipose tissue seems to matter, but a sound quantitative experimental comparison across different tissues would be more helpful. I expect in reality both the pathways could be contributing to pathology, but it is important to evaluate their relative roles since that can have important clinical implications.
There is another possible way in which the behavior-driven and glucose-driven pathways could be interacting. The hyperglycemia-driven pathways lead to oxidatively damaged proteins and cells, accumulation of AGEs, and AGE-driven protein damage. However, there is a rate at which damaged proteins and cells are being replaced in normal life. The effects of these damages need to be weighed against the rate of replacement of damaged components. In diabetes there is evidence that the rate of replacement is reduced. For example, the rate of formation of endothelial progenitor cells is reduced in diabetes [116]. This overall reduction is likely to be behavior driven since testosterone [117–119], estrogens [120–123], erythropoietin [124–129], and other behavioral signal molecules play a role in proliferation and mobilization of endothelial progenitor cells. EGF and other growth factors are needed for tissue regeneration. Testosterone, oxytocin, and some other behavioral signals promote protein synthesis. Therefore it is likely that these pathways stimulated by physical activity and aggression may effectively compensate the hyperglycemia-induced damages by replacing damaged cell components, cells, or tissues. Similarly even if we assume that hyperglycemia is the only source of oxidative radicals, antioxidant activity triggered by aggression can take care of it at least partially. Since aggression anticipates injuries and macrophages activated by injuries produce free radicals to kill bacteria, it is necessary to increase antioxidant capacity in anticipation. Therefore aggression should strengthen antioxidant defenses. Not surprisingly testosterone and estradiol have antioxidant actions [130–133], and physical exercise and aggression appears to have some testosterone-independent effect on antioxidant capacity [134–136]. If this is true it may be possible to prevent and perhaps reverse the hyperglycemia-induced pathophysiological processes by aggressive exercise even without tight glycemic control. This is a theoretical possibility currently but is testable and needs to be put to test sooner.
The two most important possible implications of the alternative pathophysiological pathways are that (1) even if raised blood sugar is demonstrated to cause some of the pathological effects, that is not sufficient to say that controlling sugar will eliminate them. Since the rise in blood sugar is associated with disinvestment from muscle and thereby loss of aggression, one would see an apparent effect of raised sugar on the behavior-driven pathophysiological mechanisms. But now, if sugar is controlled but behavior remains the same, we may not expect a mitigation of the pathophysiology. (2) If behavior is corrected appropriately, then even if sugar levels remain moderately higher, pathology can be prevented.
Partial support to the latter exists in literature on the effects of exercises. Exercises can bring about improvements in a number of functions without, before or independent of, improvement in glycemic control. Exercise increases growth factor and neurotrophin levels [137–142], improves angiogenesis [143], increases antioxidant enzymes in muscle [135, 144, 145], and enhances endothelial function [146], all independent of blood sugar levels.
The emerging alternative picture implies the need to drift away from glucocentric thinking. This is the basic inferential difference between the two models. In the orthodox model, since all pathologies are rooted in hyperglycemia, treating hyperglycemia is all that one needs to do. This is not so in the alternative picture. According to the alternative picture too, hyperglycemia can lead to pathological effects since it affects behavior and the behavioral change can result into downstream pathologies. However, in this model, controlling sugar will not mitigate complications if the behavior remains unchanged. On the other hand if the behavior is corrected or supplemented appropriately, as we will discuss in the coming chapter, pathological effects can be prevented even without normalizing glucose levels.
I have no intention to state that it is not important to control sugar, but the important point is that sole emphasis on sugar control is not sufficient. To be conservative, treatment aimed towards moderate control of glucose should continue until we have a proven alternative effective therapy against the true pathological processes of diabetes. At the same time research should focus on understanding and arresting the true pathological mediators of diabetes.
Finally we need to address the question of “incurability” of T2D here. The major depressive factor of diabetes for a patient is the understanding that he or she is a patient for life. One can never come out of diabetes. What one can do at the most is to prevent it from escalating. Unfortunately this too does not work in all cases. Glycemic control keeps on deteriorating slowly even in patients with good compliance to treatment.
A disease can be incurable for two reasons. One being that the disease causes some irreversible changes. There are many examples of this. Mammals have lost retinal regeneration capacity which lower vertebrates have. Therefore, in mammals, retinal damage is irreversible. In poliomyelitis, the viral infection can be cured but paralysis caused by the infection is irreversible and leads to lifetime disability. Is diabetes incurable because of some such irreversible changes?
It has been believed for a long time that β cell loss is irreversible. Therefore, by the popular belief, insulin resistance can be reversed, but once the progressive β cell loss begins, it cannot be reversed. However, unlike retina, we have not lost the capacity to regenerate β cells. There have been clear and reproducible demonstrations that β cells have good regeneration capacity, and this can happen by two alternative means. The β cells themselves can replicate or they can be generated from the acinar and ductal endothelial cells [147–152]. This means that as long as pancreatic tissue exists, β cells can regenerate. There is some evidence that even in autoimmune type 1 diabetes, β cells are continuously being regenerated [153, 154], although the rate of autoimmune destruction supersedes the rate of regeneration. In streptozotocin-treated rats, at least partial regeneration of β cells has been successfully brought about under the influence of EGF and gastrin [155, 156]. Therefore it is unlikely that β cell loss is irreversible. Perhaps we have failed to identify the appropriate internal environment needed for the growth of β cells. Although research on in vitro propagation of β cells for generation of functional islets from stem cells has not given us viable therapy options, it played an important role in recognizing the complex growth factor requirement for β cell growth.
Apart from EGF, a large number of other growth factors are now known to be required for β cell growth. The list may not be complete as yet, but interestingly, all the factors listed so far also have active roles in wound healing too. Insulin, IGF1, and IGF2 are important in β cell proliferation and preventing apoptosis [157–161], both IGFs are active in wound healing and also synthesized locally at the wound site [162–165]. Insulin itself has a beneficial action in wound healing [166]. Other growth factors that are involved in both β cell regeneration and wound healing are PDGF and FGF [167–169], HGF [148, 170–174], NGF [175–177], growth hormone [178–180], prolactin and placental lactogen [147, 180, 181], PTHrP [147, 182–184], gastrin [155, 156, 185], GRP [186], glucagon-like peptide [187–189], activin [190, 191], and betacellulin [148, 192]. Not only are these factors required in wound healing, most of them are synthesized systemically or locally at the site of wound after injury [172]. For the factors that are synthesized locally, some spillover in circulation is inevitable. It makes sense for the wound-healing growth factors to help β cell function since insulin itself is needed for wound healing. This raises the possibility that a chronically non-injury-prone lifestyle creates a deficiency of one or more of these factors affecting β cell regeneration. Interestingly two immunosuppressive agents that were used in the Edmonton protocol to prevent immune damage to transplanted β cells have now been shown to prevent natural β cell regeneration [150]. This is very likely to be related to the suppression of wound-healing mechanisms that the two agents are known to bring about [193–196]. Therefore the causes of impairment of wound healing and failure of effective β cell regeneration in diabetes appear to have much in common. This raises the possibility that the factors that would normalize β cell regeneration in diabetes will be identified in the near future, and a growth factor cocktail might turn out to be effective. But it is equally or perhaps more likely that behavioral intervention to stimulate growth factors and wound-healing mechanisms may work better. It is already known that two behaviors, namely, aggression and adventure, that anticipate injuries induce secretion of some of the growth factors. There are no studies on whether there is behavioral stimulation of all other growth factors, but one should expect it logically. If that is true, the right cocktail could be internally generated, and islet degeneration might be substantially reversed. If β cell loss can be reversed, then except for extremely advanced stages of diabetic complications, particularly retinopathy and nephropathy, there do not appear to be any truly irreversible changes involved in diabetes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


