Chapter 27 Anticonvulsants and Other Neurologic Therapies in Small Animals
Successful control of seizures with anticonvulsant drugs reflects a balance in achieving seizure control and minimizing undesirable drug side effects. Variability in the disposition of anticonvulsants and interactions among them and other drugs are important confounders of successful therapy. This chapter reviews selected anticonvulsants, focusing on drugs most likely to control seizures in small animals However, drugs that have been used historically and selected drugs that are used in humans but for which information is available in dogs or cats are also discussed, in part to explain why they may be less than ideal choices.The proper use of anticonvulsants is discussed, with an emphasis on the differences in individual drug disposition, detection of these differences (e.g., therapeutic drug monitoring), and rational approaches to responding to these differences by dose modification. The primary topic of discussion is treatment of generalized, tonic–clonic seizures, the most common type afflicting small animals. Opinions regarding anticonvulsant therapy vary among clinicians. Treatment of behavioral disorders that might manifest as seizural type activity is discussed in Chapter 26. Some of the comments and recommendations offered in the discussions of selected drugs reflect personal observations obtained in the direction of a therapeutic drug monitoring service or through clinical trials that focus on the use of anticonvulsants either alone or in combination with phenobarbital.
It is important to approach epilepsy as a clinical manifestation of an underlying disease. Thus therapy is more likely to be effective if the underlying disease is treated. Such causes should be identified and appropriately treated—if possible, before chronic anticonvulsant therapy is instituted. Neutering of affected animals (male and female) is strongly encouraged not only for ethical reasons (i.e., precluding perpetuation of contributing genes) but also to minimize the potential adverse effect of circulating sex hormones on neuronal membrane stability. Unfortunately, the underlying cause of epilepsy often cannot be identified (idiopathic epilepsy) or, if identified, cannot be corrected such that seizures are adequately controlled. In such instances, regardless of the cause of seizures, management is based on control with anticonvulsant drugs. Undesirable side effects are often the limiting factor in the use of anticonvulsant drugs, and not all seizures necessarily require treatment.
Physiology and Seizure Pathophysiology
Anatomy
Neurons are either layered or clustered (forming nuclei) and classified according to function (sensory, motor, or interneuron), location, or the neurotransmitter synthesized and released by the neuron.1 Neurons are outnumbered by several orders of magnitude by other cells, including macroglia, microglia, and cells of the vascular elements. Microglia are related to macrophage/monocyte lineage and are either resident or recruited during inflammation. The macroglia are the most abundant of the supportive cells and include, among other cells, astrocytes and oligodendroglia. The latter are the myelin-producing cells, whereas astrocytes provide metabolic support and remove extracellular neurotransmitters.
Barriers to Drug Distribution
The blood–brain barrier and blood–cerebrospinal fluid (CSF) barrier form a permeability boundary, rendering the brain a sanctuary by limiting penetration of macromolecules. They also act as selective barriers to inflow and outflow of small, charged molecules, including drugs, neurotransmitters, and their precursors or metabolites. The blood–brain barrier is absent in selected locations, thus allowing the brain to monitor chemicals. These areas include but are not limited to the area postrema, median eminence of hypothalamus, and the posterior pituitary and pineal glands. Cerebral ischemia and inflammation modify the integrity and function of the barriers. The blood–brain barrier comprises blood capillaries that differ in structure compared to other tissues in that they lack fenestrae and are joined by tight junctions similar to the epithelium of other tissues. Microvessels comprise about 95% of the total surface area of the barrier. Astrocyte foot processes encapsulate the capillaries, forming the tight junctions (Figure 27-1). Because pinocytosis also is absent, compounds can enter the brain only by passive diffusion, limiting penetration to small, lipid-soluble, non-ionized molecules. In addition to the physical barrier, function mechanisms exist to preclude central nervous system (CNS) penetration. These include degradative enzymes, which target not only chemicals but also many peptides, and high concentrations of efflux transporters. These include adenosine triphosphate (ATP)–binding cassette proteins such as P-glycoprotein (among the most abundant and most well characterized) and others, resulting in a combined broad substrate specificity that targets many drugs.2 The substrate specificity of the P-glycoprotein transporter is large; its role in breed susceptibility to CNS adverse drug reactions is discussed in Chapters 2 and 3. Interactions involving P-glycoprotein (or other efflux transporters) are among the drug interactions described at the level of the blood–brain barrier.3 In the dog this protein is regulated by only one gene (ABCB1).4
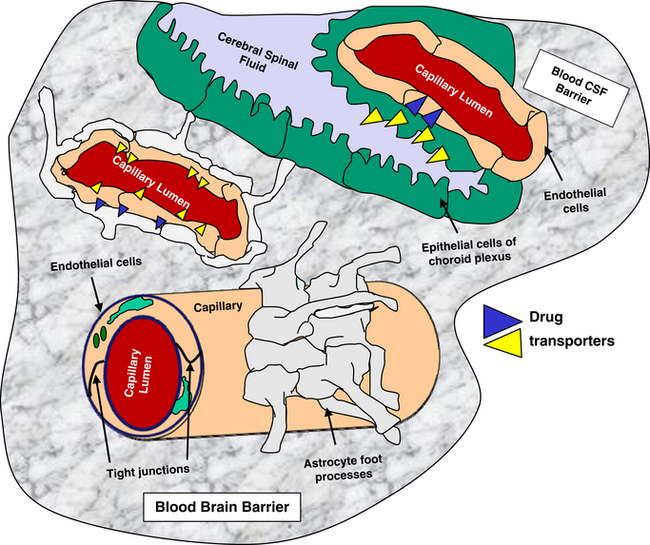
The blood–CSF barrier prevents compounds moving from interstitial fluid into brain parenchyma.5 This barrier is located in the epithelium of the choroid plexus, limiting movement of compounds from the blood into the CSF. The choroid plexus consists of the highly vascularized pia mater and cells with microvilli on the CSF side. The plexus dips into pockets formed by folding of ependymal cells onto themselves, forming a double layer structure between the dura and pia (i.e., the arachnoid membrane). This double layer contains the subarachnoid space. Although the capillaries of the choroid plexus are fenestrated, the adjacent choroidal epithelial cells form tight junctions, precluding movement of most macromolecules. Like the blood–brain barrier, the blood–CSF barrier contains organic acid transporters that preclude penetration of many therapeutic agents.6 The role of efflux pumps and P-glycoprotein in particular is discussed later.
The formidable barrier presented to chemical penetration of the brain contributes to therapeutic failure when treating dysfunction of the CNS. A number of strategies have been described to enhance drug delivery to the brain.6 These include manipulation of the drugs such that they are chemically more likely to penetrate the barrier, administration of prodrugs, the combination of drugs with compounds that decrease drug efflux, the design of vectors or other carriers that are able to penetrate, and temporary disruption of the barriers themselves. The use of nanotechnology offers potentially viable options.7 Finally, alternative routes of delivery include intraventricular, intrathecal, olfactory, and direct interstitial routes. None thus far has proved consistently safe or relevant to all therapeutic agents.
Physiology of Neuronal Activation
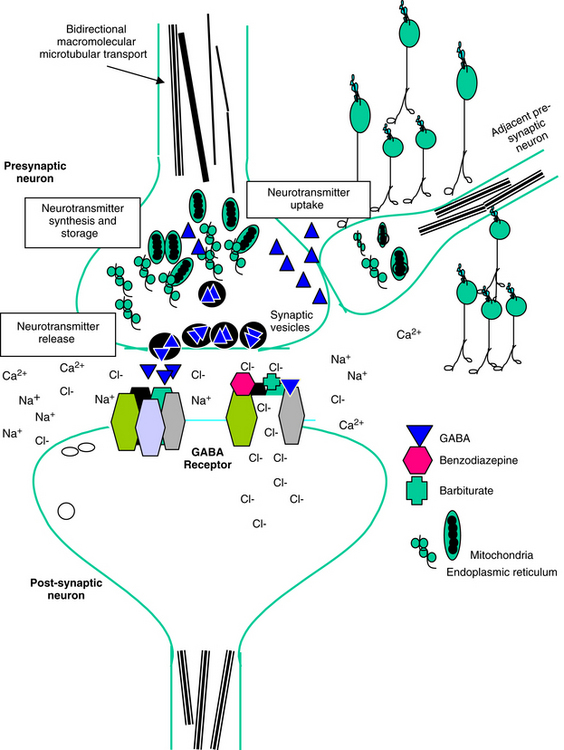
Neuronal activity can be manipulated through molecular mechanisms at several levels: (1) ion channels, which mediate excitability; (2) neurotransmitters and their receptors; (3) auxiliary intramembranous or cytoplasmic transductive molecules that couple receptor signals to intracellular actions; and (4) neurotransmitter transporters that facilitate their conservation through reaccumulation into the terminal (plasma membrane proteins) and then synaptic vesicles (vesicular transporters) (Figure 27-2). The combined influence of these molecular entities regulate three major cations (Na+, K+ and Ca2+) and the major anion (Cl–). The cations are directly regulated through voltage-dependent Ca2+, N+, and K+ channels, allowing for rapid changes in ion permeability, thus facilitating the excitation–secretion coupling necessary for transmitter release. They also are influenced by ligand-gated ion channels, which in turn are regulated by neurotransmitter binding. These include but are not limited to glutamate receptors and ionophore receptors for acetylcholine (nicotinic), GABAA, and glycine.1
Neurotransmitters
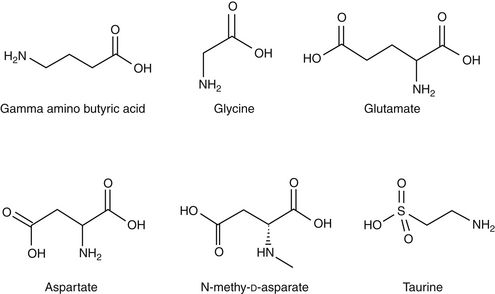
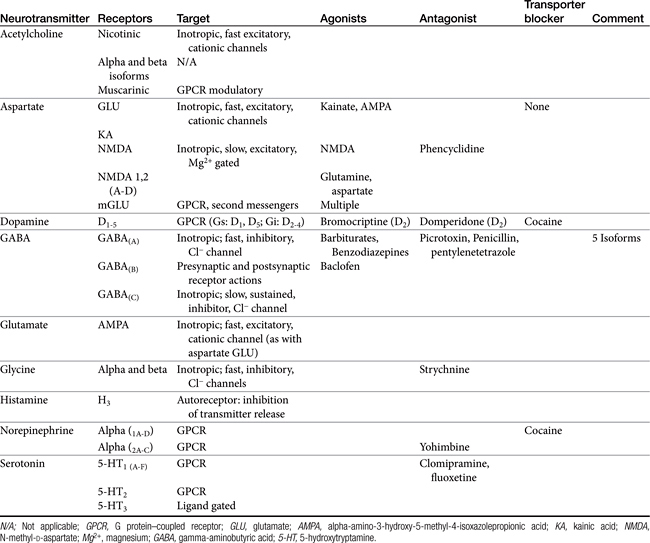
Amino acid neurotransmitters include the excitatory dicarboxylic amino acids glutamate and aspartate and the inhibitory monocarboxylic ω-amino acids glycine, gamma-aminobutyric acid (GABA), β-alanine, and taurine (Figure 27-3, Table 27-1). Because of their ubiquitous distribution in the brain and their rapid, reversible, and reduntant effects, amino acid neurotransmitters were precluded from initial inclusion as classical neurotransmitters. Glycine, glutamate, and GABA are now considered central neurotransmitters. GABA is enzymatically formed from glutamic acid by glutamic acid decarboxylase. The effects of GABA are experimentally identified in response to picrotoxin and bicuculline. These include inhibitory actions at the level of the local interneuron and potentially presynaptic inhibition in the spinal cord. Convulsant compounds that appear to act through GABA receptors include the selective antagonists penicillin and pentylenetetrazole. Nontherapeutic agents have also been identified that mimic GABA, inhibit its active reuptake, or alter its turnover. Three types of GABA receptors have been identified: A, B, and C, with type A being the most common. It is a ligand-gated, chloride channel ionotropic receptor and is the site of action of many drugs, including benzodiazepines, barbiturates, anesthetic steroids, and volatile anesthetics. The B type GABA receptor is a G protein–coupled receptor (GPCR) that interacts with Gi to inhibit adenylyl cyclase, activate K+ channels, and reduce Ca2+ conductance. The B type receptors act presynaptically as autoreceptors, inhibiting GABA release. Type C GABA receptors are less common, but GABA is much more potent for this receptor. The type C receptor is distinguished by its lack of interaction with several compounds that interact with type A GABA receptors, including baclofen, benzodiazepines, and barbiturates.
Amino Acids
Glutamate and aspartate occur in very high concentrations and are powerful excitatory signals throughout the brain. Glutamate receptors are either ligand-gated ionotropic receptor channels or G protein coupled. Ligand-gated ion channels include N-methyl-d-aspartate (NMDA) and non-NMDA receptors (alpha–amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] and kainic acid [KA]).1,8,9 The NMDA receptors, derivatives of aspartic acid, are pH sensitive, and response to endogenous signals, including zinc ions, selected neurosteroids, arachidonic acid metabolites, redox regions, and selected polyamines. Although NDMA receptors are involved in normal synaptic transmission, they appear to be more closely involved with induction of synaptic plasticity. This includes long-term potentiation, which reflects an increase in the magnitude of the postsynaptic response to presynaptic stimulation of a specific strength. The activation of NMDA receptors requires simultaneous firing of at least two neurons, including binding of glutamate released from the synapse and simultaneous postsynaptic depolarization from different neurons. High concentrations of glutamate can cause neuronal death. The mechanism partially, but not solely, reflects excessive activation of NMDA or AMPA/KA receptors and calcium influx. For example, ischemia or hypoglycemia may be associated with massive glutamate release; when coupled with impaired cellular reuptake, cell death may occur. Depletion of Na+ and K+ and increased extracellular Zn2+ may play a role in activation of necrotic or apoptotic cascades.1 The role of these receptors in pain is discussed in Chapter 28.
Monoamines
Several catecholamines act as neurotransmitters in the CNS. Dopamine comprises more than 50% of CNS catecholamine and is found in high concentrations is selected areas. Its effect depends on the subreceptor targeted, with five thus far identified in the brain (see Table 27-1). Epinephrine is present only in limited areas, and its role has not yet been identified. However, norepinephrine is found throughout the brain but in very large concentrations in the hypothalamus, certain zones in the limbic system, and other regions. All three receptor types (alpha 1, alpha 2, and beta) and subtypes are located in the brain. The nonclassical electrophysiologic synaptic effects of norepinephrine vary with the state but are considered “enabling.” Although excitatory in nature, the effects of norepinephrine (and serotonin) can be anticonvulsant, rather than proconvulsant.10
Biogenic Amines
Histamine is the second biogenic amine that serves as a neurotransmitter. Four subtypes have been described in the CNS, all acting through G proteins. Most neurons that synthesize histamine are located in the ventral posterior hypothalamus and extend throughout the entire CNS through ascending and descending tracts.1
Synaptic Vesicle Protein
The importance of the synaptic vesicle protein 2 (SV2) emerged as part of the research into the mechanism of action of levetiracetam. Levetiracetam, but not other anticonvulsants, binds to synaptic vesicle (SV) protein through the brain in a steroselective, concentration-dependent manner.11 However, ethosuximide, pentobarbital, and pentylenetetrazole compete with levetiracetam for binding. The distribution of the protein does not follow any pattern consistent with classical receptors, and the protein is located in synaptic vesicled (SVs). Three isoforms exist (A, B, C), with SV2A the most widely distributed. Knockout mice without SV2A die within 3 weeks as a result of spontaneous seizures. The role of SV2A is not clear, but it may modulate exocytosis that precedes neurotransmitter release; disruption of activity appears to result in calcium accumulation and increased neurotransmitter release.11
The Action Potential
The normal resting membrane potential (RMP) of the neuronal cell is 70 mV. The electrical difference across the cell membrane is maintained by a Na+, K+-ATPase pump. Depolarization and the generation of an action potential occur when the RMP becomes sufficiently positive to reach threshold. As with other membranes, the RMP of a neuron is determined by the concentration of negative and positive ions across the membrane. Important ions include Na+, K+, Ca2+, and Cl–. The concentration reflects ion fluxes and thus permeability of the cell membrane to the ions. Fluxes resulting in an increase in positive ions inside the cell relative to the outside hypopolarize the RMP, bringing it closer to threshold and subsequent depolarization. The tendency of a neuron to depolarize reflects, in part, the sum total effect of neurotransmitters (NTs) that interact with the cell membrane. Inhibitory NTs such as GABA render the RMP more negative and less susceptible to depolarization (see Figure 27-2).12 The principal postsynaptic receptor for GABA is the GABAA receptor; activation increases inflow of Cl- ions into the cell, hyperpolarizing the neuron. Glycine and serine are also inhibitory neurotransmitters. Excitatory neurotransmitters, such as acetylcholine and glutamate, elevate the RMP to a more positive status, rendering it more susceptible to reaching the threshold necessary for depolarization.12
Pathology
Conventional evidence indicates that neurons are terminally differentiated cells and do not respond to proliferate stimuli. The presence of neuronal stem cells may, however, offer an avenue of treatment for degenerative conditions or damage. In the absence of proliferation, neurons have developed adaptive functional and structural responses to injury.1
Several animal models have been used to study either seizures or epilepsy. Models intended to study epilepsy ideally include both seizure activity as well as chronic epileptiform behavior (i.e., spontaneous seizures).13 The attributes of animal models are generally based on their relevance to human epilepsy in regard to electrophysiologic activity, etiology, pathologies, signalment, associated behavioral changes, and response to anticonvulsant drugs. That multiple models have been developed for study reflects the more than 100 human seizure or epileptic disorders. Chemical convulsants that cause seizures after systemic or direct administration include KA (a glutamate analog), pilocarpine, NMDA, and GABA antagonists, Pentylenetetrazole is among the most commonly used chemicals; repeated injections mimic electrical kindling. Injections of high doses consistently and predictably cause tonic–clonic seizures, allowing its use as a screen for new drugs. Lower doses induce absencelike seizures. Kindling models are considered epileptic models and involve periodic stimulation of low-intensity signals in the amygdala, resulting in tonic–clonic seizures. The animal remains sensitive to electrical stimulation, with the number of stimuli necessary to induce seizures quantifiable. Genetic models have been either developed or genetically designed;13 for example, deletion of the gene encoding tyrosine kinase prevents the genesis of epilepsy in the kindling model administration of chemoconvulsants.1
Seizures are the clinical results of rapid, excessive neuronal discharge in the brain. Seizures are classified as primary (i.e., genetic) or acquired, and as generalized or focal. Generalized seizures are much more common in small animals; the incidence is greater in dogs than in cats. With seizure onset of a generalized character, convulsive electroencephalographic activity begins simultaneously in all brain regions.14 Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate seizures and by the neurobiologic, cognitive, psychological, and social consequences of this condition. The definition of epilepsy requires the occurrence of at least one epileptic seizure.10
Seizures in epileptic subjects often are attributed to a cortical origin. However, the brainstem, particularly the pontine reticular formation, also can manifest self-sustained seizure discharge and may be important in the generation and expression of generalized tonic convulsions.15 Depression of reticular core activity is an essential characteristic of antiepileptic drugs, suggesting that the reticular formation is involved in the spread and generalization of clinical seizures.16 In the dog the most common form of epilepsy is generalized tonic–clonic or grand mal seizures.17 Epilepsy or seizure disorders of the CNS in the dog may be caused by an acquired organic lesion such as brain tumor, head trauma, toxicosis, electrolyte imbalance, hypoglycemia, renal failure, or hepatic disease (acquired or secondary epilepsy) or may be genetic or inherited (“true,” idiopathic, or primary epilepsy). An autosomal gene associated with a sex-linked suppressor on the X chromosome may explain the higher incidence of seizures in male dogs. Interestingly, in human medicine the role of autoantibodies is emerging as a potential underlying cause of neurotoxicity and associated epileptic seizures.18
Status epilepticus (SE) refers to failure of the patient to recover to a normal alert state between repeated tonic–clonic attacks or episodes that last at least 30 minutes.19 Convulsive or tonic–clonic SE is a medical emergency in which convulsive seizures must be terminated by treatment with anticonvulsant agents. In humans epileptic seizures must not be allowed to persist more than 60 minutes if severe, permanent neurologic injury or death is to be avoided.19 The longer an epileptic seizure persists, the greater the incidence of mortality and morbidity. Hyperthermia caused by continuous muscle contraction may become life threatening during continued seizure activity. Brain damage resulting from hypoxia or the sequelae of hyperthermia is more likely if more than 30 minutes of uninterrupted seizures occur.
During seizures individual neurons depolarize and fire action potentials at high frequencies. Increased excitability reflects increased extracellular potassium and decreased extracellular calcium. Once initiated, the seizure discharge may synchronize with other neurons and propagate or spread to surrounding areas in the brain. Seizures can be initiated by four general mechanisms:
Principles of Anticonvulsant Therapy
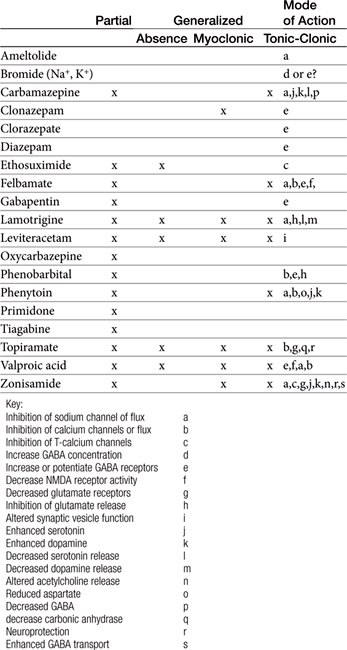
Anticonvulsants block seizure initiation and propagation by blocking either abnormal events in a single neuron or the synchronization of related neurons. Thus a goal of therapy is reduction in the firing frequency. A common mechanism by which this goal might be achieved is prevention of sodium channel recovery from inactivation (i.e., prolongation of the refractory period) (Table 27-2). It is during this period that membranes are nonresponsive to other signals. Example drugs that target this mechanism include carbamazepine, phenytoin, valproic acid, toparimate, and zonisamide.25 Another common mechanism is increased interaction between agonists and GABAA, which facilitates hyperpolarization. Drugs that act as agonists at the GABA(A) receptor include the barbiturates, and the benzodiazepines. At high concentrations these drugs also may also inhibit high-frequency firing. Other drugs include tiagabine, which targets the GABA transporter, GAT-1, decreasing neuronal and glial uptake of GABA and levetiracetam, which targets SV proteins. In general, drugs that target more than one mechanism tend to be most effective. Alternatively, combination therapy with drugs that target different mechanisms of neuronal function may be effective in seizures that do not respond to single-drug therapy.
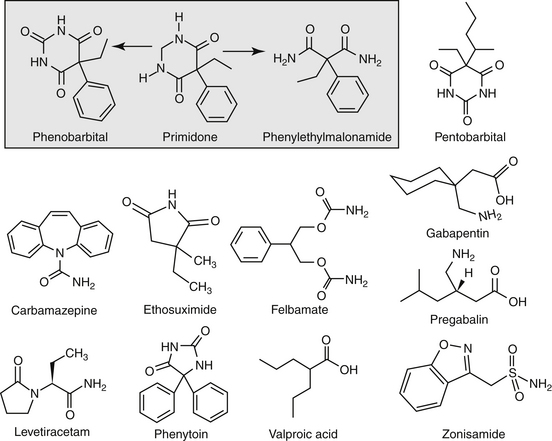
Anticonvulsant drugs target seizures; antiepileptic drugs target epilepsy. For the remainder of this chapter, the terms may be used interchangeably, in part because most antiepileptic drugs are also anticonvulsant drugs. Because successful anticonvulsant therapy may depend on the maintenance of plasma drug concentrations within the therapeutic range, understanding the disposition of each anticonvulsant is paramount to therapeutic success.26,27 Epilepsy is controlled, not cured, with historical rates of success in only 60% to 70% of the cases, although control might be improved to at least 85% if monitoring supports therapy (see later discussion).28 Generally, but not always, treatment for epilepsy involves drug administration for the rest of the animal’s life.29 The most common anticonvulsant drugs that have been used in veterinary medicine are phenobarbital, diazepam, and potassium bromide (Figure 27-4). A number of newer (human) drugs are increasingly being used, including zonisamide, levetiracetam, several benzodiazepines (clorazepate, clonazepam), gabapentin, and (less frequently) felbamate. Older drugs whose historical use has declined include primidone, phenytoin, and valproic acid. Drugs commonly used in humans that are minimally used in dogs or cats include ethosuximide, lamotrigine, and topiramate. Scientific support of efficacy for each of these drugs in dogs or cats generally is limited; differences compared with humans should be anticipated, not only because of differences in the underlying pathophysiology of disease but also in the disposition of the drugs among species and pharmacodynamic response. Even if pharmacokinetic information is available, generally the data were generated in a small sample size of healthy, drug-free animals. Pharmacokinetic differences in anticonvulsant drugs among and within breeds and species can be profound, with drug disposition often affecting the efficacy of the drug. Studies in Beagles or other pure breeds may not reflect the canine population as a whole. Antiepileptic drugs are generally administered chronically. As such an understanding of the determinants of drug disposition (Chapter 1) is paramount to understanding their most effective use, particularly as it pertains to drug toxicity and avoidance of drug fluctuations during a dosing interval that might contribute to either therapeutic failure. The more important points are as follows.
Pharmacokinetic Considerations
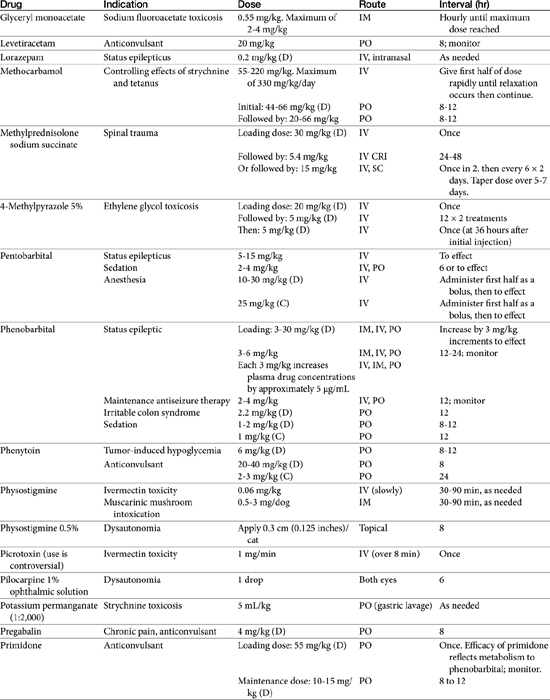
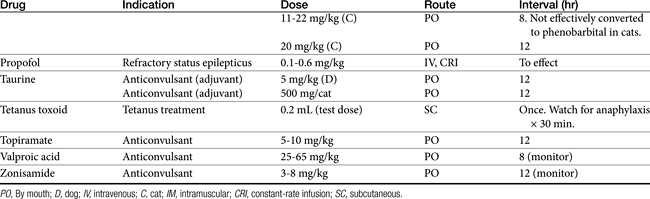
Absorption determines the magnitude of, but also the time to, peak anticonvulsant effect. Most anticonvulsants are administered either orally or intravenously, although alternative routes such as nasal and rectal administration are proving increasingly useful for emergency administration (Table 27-3). Most of the anticonvulsants used as antiepileptic drugs are well absorbed after oral administration, with the notable exception of phenytoin. Because food may slow the absorption of anticonvulsant drugs, peak plasma drug concentrations may occur as late as 4 to 6 hours after administration. This generally is not a problem with chronic dosing for drugs that accumulate, but it may affect the time of peak sample collection. Fasting before sample collection might be prudent. Rectal administration has offered a clinically effective alternative for administration for several anticonvulsant drugs. The colonic absorption of anticonvulsant drugs can be predicted to some degree by hydrophilicity. For example, whereas gabapentin is poorly bioavailable compared with phenytoin, maximum plasma concentration of phenytoin after colonic administration equals that after oral administration.30 A potentially emerging problem — suggested by the author’s therapeutic drug monitoring service — are differences in oral bioavailability of human generic products prescribed for animals. The therapeutic equivalence (including bioavailability) that characterizes human generic products may not translate to therapeutic equivalence when that product is used in animals. Pharmacies may change a generic product intermittently as product prices change. Dispensing pharmacists may not understand the potential risk of differences in bioavailability between animals and humans. The prudent client should query pharmacists regarding changes in products, and the discerning clinician might encourage monitoring with new prescriptions, particularly in patients at risk for life-threatening seizures.
KEY POINT 27-6
Bioequivalence required of human drugs may not translate to bioequivalence in animals.
Distribution into the CNS is important for all anticonvulsant drugs, although at steady state, each generally distributes sufficiently into the CNS. Indeed, most anticonvulsant drugs are sufficiently lipid soluble that they are distributed to a volume that exceeds total body water (i.e., more than 0.6 L/kg). In contrast to chronic use, for acute situations rate and amount of drug movement into the CNS may not be sufficient for the rapid response necessary to resolve life-threatening seizures. Drug binding to serum proteins may limit the amount and rate of drug moving into the CNS. However, diazepam is the most lipid-soluble anticonvulsant and very rapidly distributes in the CNS despite its 90% protein binding (see Figure 27-4). Phenobarbital is less lipid soluble and is 50% protein-bound, and therapeutic effects may take as long as 15 minutes to be achieved. Löscher and Frey31 described the relative half-times to penetration of anticonvulsant drugs into the CSF of dogs. For diazepam its active metabolites desmethyldiazepam and oxazepam, clonazepam, and ethosuximide, the distribution half-time was 3 to 7 minutes, compared with 12 to 18 minutes for valproic acid, phenytoin, phenobarbital, and carbamazepine. In contrast, distribution half-time for primidone, its metabolite phenylethylmalonamide, and the active metabolite of carbamazepine (i.e., carbamazepine-10, 11-epoxide) was 40 to 50 minutes. Lipophilicity, rather than extent of protein binding or degree of ionization, was the major determinant of the rate of distribution.
The role of P-glycoprotein and other ABC transporters in CNS drug distribution is well recognized, being present at both the blood–brain and blood–CSF barriers. The efflux pumps, often accompanied by drug-metabolizing enzymes, contribute to the poor CNS drug penetrability. However, Mealey and coworkers32 suggest that the impact of P-glycoprotein is very limited at the blood–CSF barrier compared with the blood–brain barrier in dogs, based on similar penetration of a radiolabeled P-glycoprotein substrate into the CSF, but not brain, of normal (wild-type) dogs compared with ABCB1 knockout dogs. The role that efflux pumps contribute toward epilepsy, particularly refractory, is emerging.2 Increased concentrations of Ppg are associated with seizures and refractory in human patients. In animal models of refractory epilepsy, efflux pump inhibitors have been associated with an increase in drug response. Yet several studies have demonstrated that the anticonvulsant drugs often are not substrates for Ppg. Using a canine osteosarcoma cell line, West and Mealey4 demonstrated that diazepam, gabapentin, lamotrigine, levetiracetam, and phenobarbital were weak substrates, whereas carbamazepine, felbamate, phenytoin, topiramate, and zonisamide were not substrates. However, other factors may affect the state of P-glycoprotein. For example, glutamate induces P-glycoprotein expression, which may contribute to a pre-epilpetic condition. Pekcec and coworkers33 demonstrated upregulation of P-glycoprotein in dogs after an episode of SE. Furthermore, a number of other transporters may exist in the brain that might also affect anticonvulsant drugs.
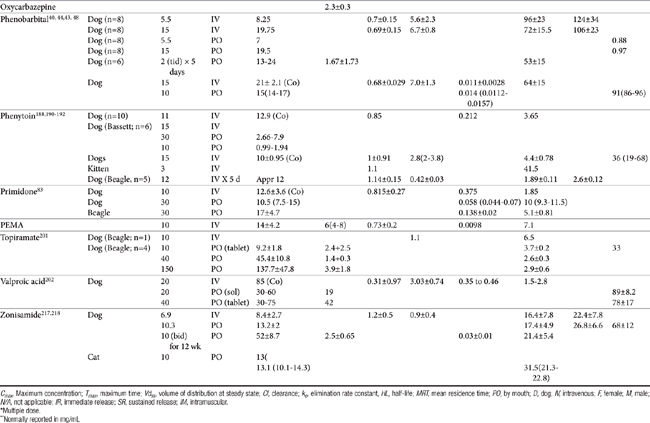
Lipid solubility, which characterizes many anticonvulsants, necessitates hepatic metabolism if the drugs are to be eliminated. Metabolism of anticonvulsant drugs can have a profound effect on therapeutic success. The effect in part depends on the sequelae of phase I metabolism on the particular drug (i.e., inactivation, activation, or generation of toxic compounds). The rate of metabolism of anticonvulsants is variable: Phenobarbital is slowly metabolized, characterized by a long half-life, whereas diazepam is metabolized rapidly and is characterized by a short half-life (Table 27-4). The rate of metabolism may vary markedly among species: Phenytoin is metabolized slowly in humans but is characterized by a half-life of less than 2 hours in dogs. Phase I metabolism often yields an active compound; anticonvulsant efficacy of the metabolite may be equal, greater, or less than that of to the parent drug. Primidone must be metabolized to its active metabolite, phenobarbital before it is effective in dogs. Clorazepate, a benzodiazepine, is a prodrug derivative of a diazepam metabolite that is converted in the stomach to its active form (Figure 27-5). Although diazepam is rapidly metabolized, its duration of pharmacologic effect is prolonged because most of its metabolites have some degree of anticonvulsant effect. The half-life of the metabolites may also be longer than those of the parent compounds.
Hepatic metabolism affects both efficacy and safety, in part because of its influence on fluctuation of drug concentrations during the dosing interval. Although it is clearance (regardless of hepatic or renal) that physiologically affects drug concentrations throughout the dosing interval, half-life is the practical pharmacokinetic measure of its impact (Box 27-1; see also Chapter 1). Neither antiepileptic efficacy nor safety of an anticonvulsant drug should be evaluated until at least 87% of steady-state concentrations has been reached, plus one seizure interval (such that the brain is challenged; see Table 27-1). Every time a dosing regimen is changed, a new steady-state equilibrium should be reached before final reevaluation. The time that must elapse before steady-state concentrations and maximum therapeutic response can occur may be unacceptably long for patients suffering from severe, life-threatening seizures or from unacceptable adverse effects to the initial antiepileptic drug. For such patients a loading dose might be administered such that therapeutic antiepileptic drug concentrations are achieved rapidly. The amount of the loading dose necessary to achieve steady-state concentrations (not equilibrium) increases proportionately with the half-life of the drug to be loaded: The loading dose is a sum of all the daily doses that would have been administered before steady state occurs, less any drug that will be eliminated from the body during that time period. The major disadvantage of a loading dose is the sudden effect of therapeutic concentrations in the CNS; no time is allowed for adaptation, and adverse effects (sedation, ataxia) are more likely than with gradual increases in drug concentrations. The maintenance antiepileptic drug dose that follows the loading dose is designed to maintain concentrations achieved by the loading dose. Both loading and maintenance doses are based on population disposition parameters; monitoring should be used to ensure that dosing regimens achieve targets for the patient. Generally, antiepileptic drug monitoring is indicated immediately after the loading dose is completed (e.g., the next day). Patients should be monitored again one half-life later to ensure that the maintenance dose is able to maintain concentrations achieved with the loading dose. If the maintenance dose does not maintain what is achieved with the loading dose, the majority of the change as new steady state is reached with the new dose will occur during the first half-life of the drug.
Safety
The safety of anticonvulsant drugs is profoundly affected by metabolism, not only by virtue of short elimination half-lives (for some drugs) but also because of toxicity. Phase I metabolites, by their nature, are reactive. Although intended to progress to phase II metabolism (with subsequent detoxification, particularly by glutathione), some reactive metabolites can interact with and damage surrounding tissues; concentration in the liver can lead to predictable (type A or type I, also referred to as “intrinsic”) hepatotoxicity. Thus the risk of hepatotoxicity is likely to be increased with dose and frequency of administration.However, idiosyncratic toxicity (i.e., neither dose nor duration dependent) may also be the sequelae of a metabolite. Although dogs receiving chronic anticonvulsant therapy often develop abnormalities in serum biochemistries and hepatic function tests,34–36 only about 15% of dogs receiving long-term anticonvulsant therapy have been estimated to be at risk to develop serious hepatotoxicity. This risk is, however, greatly increased if drug concentrations approach the maximum therapeutic range. Primidone is probably most commonly associated with hepatotoxicity in dogs, followed by phenobarbital and then phenytoin in combination therapy.34–36 One of the many reasons that phenytoin is no longer used as an anticonvulsant in dogs is hepatotoxicity. The risk of hepatotoxicity is discussed with each individual anticonvulsant. Toxicity may be enhanced by concurrent administration of drugs (e.g., phenobarbital) that induce drug-metabolizing enzymes and therefore increase the formation of potentially toxic (particularly phenytoin) intermediates.
Selected anticonvulsants (including phenytoin, phenobarbital, and zonisamide) can affect thyroid hormones; testing before initiation of anticonvulsant therapy might be prudent. Mechanisms include displacement from binding proteins (e.g., phenytoin), increased peripheral metabolism (e.g., phenobarbital), or decreased synthesis (e.g., zonisamide).These effects are discussed with the individual drug. Several anticonvulsants have been associated with changes in serum lipids (e.g., phenobarbital, carbamazepine, valproic acid) in both dogs37 and humans.38
Drug Interactions
Most drug interactions involving antiepileptic drugs occur at the level of hepatic drug-metabolizing enzymes. Hepatic metabolism increases the risk of drug interactions; use of any drug metabolized by the liver should be lead to a focus on potential drug interactions. For some anticonvulsants (especially phenobarbital), drug interactions and sequelae have been well described; both induction and inhibition of drug-metabolizing enzymes have been reported. Phenobarbital is the most potent hepatic drug-metabolizing enzyme inducer known and will increase the rate and extend of hepatic metabolism of many drugs, including phenobarbital. Combinations of anticonvulsants can lead to unpredictable effects on drug metabolism. Antiepileptic drugs metabolized by the liver likewise can be affected by other drugs that alter drug-metabolizing enzyme. Cimetidine, ketoconazole, and chloramphenicol are drugs that decrease drug metabolism and have the potential to profoundly increase concentrations of the anticonvulsant. Drug interactions may also involve P-glycoprotein or other efflux transporters.2 The impact of these interactions is an emerging area of research for antiepileptic drugs.
Therapeutic Drug Monitoring
Therapeutic drug monitoring can be a critically impotant tool in the successful control of the difficult epileptic patient. However, not all anticonvulsant drugs can be nor need be monitored (see Chapter 5). For example, a therapeutic range must be established for the drug (and, if established in humans, be relevant for dogs or cats), and response must correlate with plasma drug concentrations. An easy, cost-effective assay that requires minimal sample handling must be available. Among the anticonvulsants to be discussed, automated assays are available for several anticonvulsants whereas more tedious and thus costly assays (e.g., high performance liquid chromoatography) must be implemented for others. Regardless of the methodology,
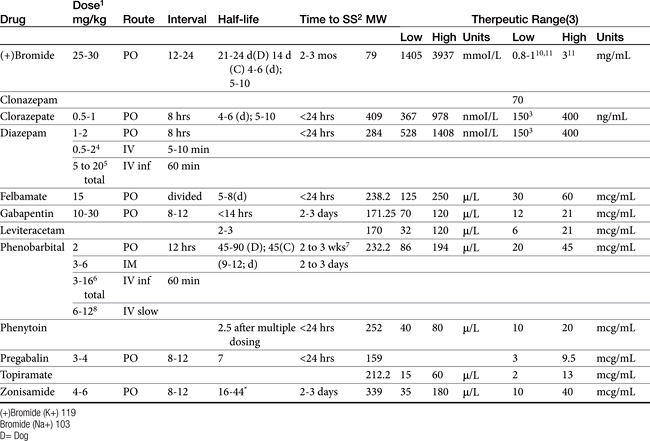
In human medicine, the use of therapeutic drug monitoring for newer anticonvulsants (which tend to be safer) is not as well established compared with that for older drugs, in part because of the lack of generally accepted target ranges (Table 27-5). However, Bialer and coworkers27 demonstrated a concentration–response relationship for antiepileptic drugs in humans, justifying monitoring. Controlled clinical trials establishing concentration–response relationships are needed in both human and animals to establish therapeutic ranges. Currently, the range in serum concentration associated with efficacy is large for some drugs and overlaps with toxicity in some patients.39 Nevertheless, therapeutic drug monitoring remains a recognized tool for individualization of drug therapy. The author suggests a “start-up”, “follow-up”, “check-up,” and “what’s-up” approach to monitoring (Box 27-2). Clearly, monitoring should be implemented to avoid toxic concentrations and to establish the minimum effective range for the epileptic patient This latter reason is particularly relevant for previously controlled patients suffering from breakthrough seizures (previously well controlled): a decrease in drug concentrations compared with baseline should lead to a focus on owner compliance, drug interactions, changes in disposition, and so forth, whereas seizures despite no change in concentrations might be interpreted as worsening of underlying disease and the potential need to control seizures more aggressively. therapy not be considered failed until trough concentrations are in the higher end of the therapeutic range. Serum separator tubes should not be used for collection of samples intended for therapeutic drug monitoring.
Box 27-2 Therapeutic Drug Monitoring and Antiepileptic Drugs
Other considerations: Animals should be fasted before samples are collected, particularly for drugs that do not accumulate (i.e., those that are characterized by a short half-life). Special handling preparations generally are unusual, although each laboratory should be called to confirm special handling procedures. Serum separator tubes are contraindicated because the silicon separator can bind anticonvulsant drugs and falsely decrease serum drug concentrations. If used, serum should be immediately harvested after centrifugation.149
Anticonvulsant Drugs
Phenobarbital
Mechanism of Action
Phenobarbital sodium (see Figure 27-4) specifically depresses the motor centers of the cerebral cortex, thus enhancing anticonvulsant properties. Electroshock experiments with cats and other species established phenobarbital as one of the most potent anticonvulsants available. It has the widest spectrum of activity in different convulsive seizure patterns. Many AED have been synthesized as structural variants of phenobarbital. For example, primidone is a close congener of phenobarbital.
Phenobarbital is the most effective anticonvulsant to delay progressive intensification of seizure activity that may accompany epilepsy. Phenobarbital both increases the seizure threshold required for seizure discharge and decreases the spread of discharge to surrounding neurons; its primary mechanism is enhancement of responsiveness to the inhibitory postsynaptic effects of GABA. Interaction of GABA opens a chloride channel, resulting in higher intracellular concentrations of chloride and hyperpolarization of the RMP (see Figure 27-3). Phenobarbital also, however, inhibits glutamate activity and probably calcium fluxes across the neuronal membrane. A study that compared CSF concentrations of GABA and glutamate in epileptic Labrador Retrievers versus non–Labrador Retrievers and in non-epileptic Beagles also compared concentrations in those epileptic dogs treated with phenobarbital and those not treated.22 Within the non–Labrador Retriever phenobarbital-treated group, GABA, glutamate, and aspartate CSF concentrations were lower compared with those of their untreated counterparts. This difference was not detected in the Labrador Retrievers treated versus untreated groups; however, only 14 Labrador Retrievers were receiving phenobarbital, which may have limited the power to detect a significant difference.
Phenobarbital might be considered a broad-spectrum anticonvulsant. Despite introduction of new antiepileptic drugs, phenobarbital has generally been the anticonvulsant of choice for the cat and dog.17 It is effective in all types of epileptic seizures observed in cats and dogs.
Disposition
As a weak acid (pKa 7.3), phenobarbital is well absorbed after oral administration, although peak plasma concentrations may not be reached for 4 to 6 hours after administration. The absorption half-life in dogs is 1.27 ± 0.21 hours.40 With an elimination half-life of 0.26 ± 0.18, about 6.4 hours may be required for near complete absorption of phenobarbital from the gastrointestinal tract. Absorption is 88% to 95% complete.41 Phenobarbital is 45% bound to serum protein in dogs.42 The volume of distribution of phenobarbital in dogs is 0.7 ± 0.15 L/kg and 0.7 ± 0.4 in cats.43 Approximately 16 days (8 to 15.5 days) of multiple dosing is necessary to attain steady-state serum concentrations in the normal (uninduced) animal; however, induction may cause this period to be shorter after a change in the dosing regimen of an animal already receiving phenobarbital. Maintenance doses of 5.5 mg/kg daily (in one to three equal doses) administered orally are required to reach an average serum concentration of 20 μg/mL, according to one study.44
Breed differences may exist in phenobarbital clearance. Frey and Löscher42 reported a half-life of phenobarbital at 64 ± 15 hours in largely mongrel dogs, but 32 ± 4.8 in Beagles; clearance was 7.0 ± 1.3 and 13 ± 1.7 mL/kg/min, respectively. Alkalinization of urine (pH >7.5) accelerates excretion of unaltered phenobarbital by reducing back-diffusion (tubular reabsorption) through drug ionization.45 Fukunaga and coworkers46 demonstrated that a twice-daily fed diet containing either 0.6 gof potassium citrate (pH 7.8 to 8.2) or 0.2 g of ammonium chloride (pH approximating 5.9 to 6.5) increased urine clearance of a single dose of phenobarbital compared with a water vehicle (pH 6.8 to 7.5) in Beagles (n = 5; female). However, the effect did not significantly (statistically nor clinically)impact peak plasma drug concentrations (μg/mL: 3.25, 2.96, and 2.97 μg/mL for water placebo, ammonium chloride, or potassium citrate, respectively) or area under the curve. Yet, half-life was affected (52, 59.6, and 51.1 hours, respectively), and as such peak concentrations might be slightly affected if the drug is dosed to steady state.
Phenobarbital is a potent inducer of hepatic drug-metabolizing enzymes and is capable of increasing the rate of clearance of other drugs metabolized by the liver as well as increasing its own rate of metabolism.47 In the dog phenobarbital (2 mg/kg) administered orally 3 times a day for 5 days results in an elimination half-life between 37 and 75 hours, with a mean elimination half-life of 53 ± 15 hours.44 After a single 5-mg/kg intravenous dose, clearance is 5.6 to 6.6 mL/kg per hour, and elimination half-life is 92.6 ± 23.7 hours in dogs40 and 47 ± 3 hours after oral administration (5 mg/kg) in cats. The effects of multiple doses of phenobarbital were documented by Ravis and coworkers.48 After 90 days of treatment (5.5 mg/kg), mean elimination half-life decreased from 88.7 ± 19.6 hours to 47.5 ± 10.7 hours in dogs. In cats 21 days of oral phenobarbital at 5 mg/kg resulted in a half-life of 43 ± 3 hours.43
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


