8 Noninfectious Respiratory Problems
Case 8-2 Premature Lungs in a Foal from an Emergency C-Section
’03 Surf’s Up arrived into the world at 305 days of gestation, 35 days before her due date (Figure 8-1). A Thoroughbred with gilded bloodlines, she had been destined for great things, and there was no doubt about her breeding date. The night watchman found her in the stall early that morning. It was a good thing, because she would not have survived for very long in the chill of upstate New York in February. The farm veterinarian had been worried that the foal might arrive early, as the mare had developed signs of placentitis at eight months of gestation. The mare had been treated with trimethoprim-sulfa and altrenogest since that time, but no one thought the foal would arrive this early. The farm veterinarian didn’t hesitate in deciding to send the foal to a referral hospital. She quickly administered colostrum via nasogastric tube, as well as 5% dextrose, penicillin, and amikacin intravenously, and sent the foal on her way.
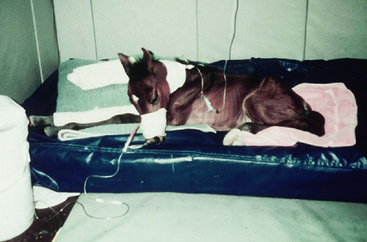
The foal was small, and somewhat weak, but his ears were erect and his fetlocks appeared only mildly hyperextended. They dried him off, brought him to the barn, and prepared to rear an orphan foal. He seemed to have a strong suckle, and eagerly drank first the banked colostrum and then the milk replacer that was offered. He stood with difficulty in six hours, and when he did, his fetlocks sank almost to the ground. He tired quickly. The owners were happy to sleep in the barn in order to feed the foal every two hours, and the veterinarian agreed to see them first thing in the morning. When the veterinarian arrived, she found a very different foal. He was recumbent, weak, and trembling. His nostrils flared with every breath, and his body rocked with the effort of his breathing. He grunted audibly at the end of each expiration. The owners reported that he had been eager to nurse until about two in the morning, when he appeared tired. He had not gotten up again.
Upon arrival at Tufts at 16 hours of age, his temperature was 97.0° F, heart rate was 160 beats/minute, and respiratory rate was 65 beats/minute. The foal’s mucus membranes were muddy, and capillary refill time was >3 seconds. His respiratory effort had not improved (Figure 8-2).
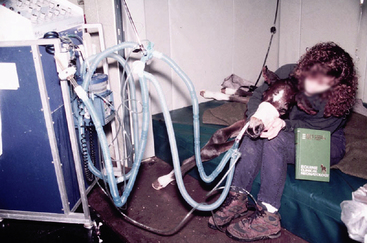
HISTORY AND PHYSICAL EXAMINATION
’03 Surf’s Up and ’03 Last Gasp were chosen for this chapter to introduce the concepts of prematurity, readiness for birth, maternal stress, and the effects of these states on the maturation of the pulmonary system of the equine fetus/foal. Traditionally, a premature foal has been defined as being born before 320 days of gestation. However, the gestation period of the equid is markedly variable (320 to 360 days).1 Some foals may be surprisingly mature at 320 days of gestation, while others meant to be born at 360 days of gestation but taken earlier, may be premature at 330 days of gestation. A syndrome of dysmaturity, wherein the foal is recognized to have the attributes of prematurity despite having achieved a gestational age of >320 days, is also recognized.2
Foals that experience “maternal stress,” in utero, such as placentitis or twinning, appear to have accelerated maturation of their pulmonary system. This is also true in human pregnancies. These foals are “ready for birth” at an early gestational age and actually signal the parturition process. “Readiness for birth” is a term coined by Rossdale that denotes signaling from the foal to produce parturition.2 These foals may have some respiratory clinical signs of immature lungs or mild pulmonary hypertension from persistent fetal circulation.
Another way to look at this is that failure of the lungs to mature by the time of birth in foals can be considered of endogenous or exogenous origin. The endogenously premature foal is one that has experienced considerable physiological intrauterine stress, resulting in a uterine environment that is no longer capable of supporting the foal. These foals are generally born before day 320 of gestation. There is often a history of placentitis (as was the case with ’03 Surf’s Up), twinning, or other systemic disease.3 The exogenously premature foal, on the other hand, is one that has had a completely normal gestation up to the time of parturition: an external event, such as a C-section mandated by acute trauma to the mare or early induction, determines the early parturition. The exogenously premature foal may be born after 320 days, and must be considered premature regardless of earlier definitions of prematurity (i.e., earlier than 320 days).
The obvious outward attributes of prematurity are those that indicate that even if the foal has reached the requisite 320 days, it is functionally premature; these foals are generally small, their ears are floppy, their carpi and tarsi are often lax due to insufficient cartilage development, and their hair coats are short and silky.4 The most important and alarming finding on physical examination of the premature foal, however, is difficult breathing. Both endogenously and exogenously premature foals may show early signs of respiratory system impairment, ranging from nostril flare to paradoxical breathing at birth; in others, subtle nostril flare is merely a precursor for subsequent respiratory failure. The exogenously premature foal is far more likely to lapse early into overt respiratory failure, while the endogenously premature foal more frequently avoids this scenario.2
As seen in Chapter 6, physical examination of the respiratory system in foals can be fraught with inconsistencies. Auscultation of the lungs can be normal despite widespread consolidation. Abnormal crackles and wheezes may be normal in the foal immediately after birth as the foal takes its first breaths and dissipates the residual fluid in the lung. If a recumbent foal is in lateral recumbency, then abnormal fluid sounds are often heard in the down lung due to atelectasis. Respiratory rate can be elevated for several nonpulmonary reasons as well. Excitement and acidosis can be causes of hypernea. The most reliable sign of respiratory problems in the equine neonate is increased respiratory effort. The foal with pronounced nasal flare, marked abdominal effort, expiratory grunting, or the appearance of paradoxical breathing is in respiratory distress.
In the case of the foal in respiratory distress, the need for immediate treatment must outweigh the desire for a meticulous physical examination and pursuit of clinical laboratory data. Cyanosis is often absent in the presence of severe hypoxemia,5 so the appearance of pink mucous membranes alone should not lead the clinician to the conclusion that the respiratory system is functioning properly. Even in the case of apparent eupnea, it is important nonetheless to obtain an arterial blood gas. Koterba found that although only 4 out of 38 septic foals in one study appeared to be in respiratory distress, 83% were hypoxemic.6 If there is more than one able hand present, one person may obtain a sample for arterial blood gas analysis while the other begins nasal insufflation of oxygen. As long as the foal is breathing, nasal insufflation of oxygen is usually sufficient for preliminary resuscitation. At this point, the careful physical examination may commence.
PATHOGENESIS
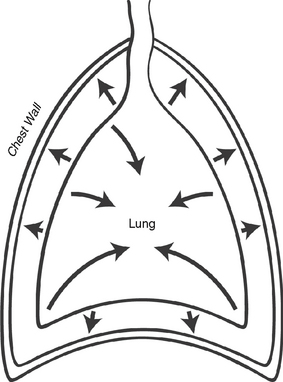
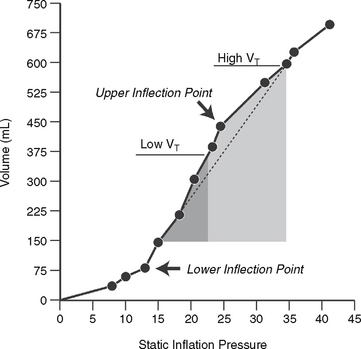
In both the normal and abnormal foal, the tendency of the lung to collapse is counteracted by the tendency of the chest wall to spring outward (Figure 8-3). The meeting place of the two determines the functional residual capacity (FRC), which is the natural resting place of the lung.7 The FRC acts as an important reservoir of air, helps to prevent collapse of the lung with each breath, and is critical in preventing the very high work of breathing that accompanies cyclic lung collapse. We can see this on a quasi-static pressure volume (PV) curve (Figure 8-4). Picture the lung at residual volume—the very smallest volume that the lung can achieve. If we then inflate the lung by increments, we see that at the earliest portion of the inflation curve, increasingly high pressures are needed to elicit a change in volume. At the end of this portion of the curve, the PV curve becomes relatively linear. The beginning of this linear phase is known as the lower inflection point (LIP), and represents the point at which collapsed alveoli begin to open.7 Breathing below the LIP requires a high level of work of breathing—neonates should not have to repeat this work after the first postpartum breaths. The premature neonate, lacking a relatively stiff chest wall to counteract the inward elastic recoil of the lung, must employ other strategies to preserve its FRC.8 One such strategy is to breathe against a closed glottis, which allows the foal to trap air at end-expiration, or “auto-PEEP,” (positive end expiratory pressure) and also elicits the characteristic grunt often heard from foals in respiratory distress. Although the neonate of all species has higher chest wall compliance than the adult, this difference is far less in the normal neonatal foal, which has an impressively stiff chest wall in comparison to other neonates. This helps the neonatal foal to preserve its FRC.9
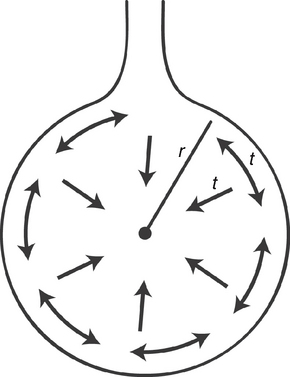
The physics of the chest wall really pale in comparison to the physics of the alveoli and surfactant. Indeed, surfactant is the sine qua non of the respiratory system. A sufficient understanding of the Laplace relationship: P = 2T/R (where P = pressure within the alveolus, T = surface tension of the alveolus, and R = the radius) helps to clarify the situation. This equation implies that if surface tension is not allowed to vary, then as radius decreases (as with a small alveolus), pressure within the alveolus becomes linearly greater. Imagine that an alveolus is attached to a second alveolus of twice the radius; the pressure in the larger alveolus will be one half that of the smaller alveolus. Thus, to equalize the pressure, the air from the smaller alveolus will flow into the larger alveolus (Figure 8-5). Thus, the natural tendency of the lung is to allow the smaller alveoli to collapse in favor of emptying into larger alveoli, with an overall decrease in surface area and volume of the lung.10
Now picture the neonate with insufficiently developed lungs. The chest wall alone would predispose the premature foal to respiratory failure; the less developed, more compliant chest wall of the premature foal, combined with muscle weakness, will inevitably result in a decreased FRC as inward recoil of the lung overcomes the outward spring of the chest wall, resulting in difficult breathing.8 It would seem that the Laplace relationship, in conjunction with the high compliance of the neonatal chest wall, would result in a lung that continually returns to residual volume even in the term foal, thus entailing enormous work of breathing with every breath.10 It is important to know that surfactant turns the Laplace relationship topsy-turvy. It not only reduces surface tension at the air-liquid interface in the alveolus, but it allows this surface tension to vary with the diameter of the alveolus.
WHAT IS SURFACTANT, AND WHY IS IT SO IMPORTANT IN PRETERM FOALS?
Pattle first described the stability of pulmonary foam,11 and Avery and Mead soon discovered that surfactant deficiency was the cause of hyaline membrane disease in premature infants.12 The majority of surfactant is a combination of phospholipids, the most abundant of which is phosphatidylcholine (PC), or lecithin. Approximately 50% of PC is saturated, or dipalmitoylphosphatidylcholine, known as DPPC. DPPC is most critical for decreased surface tension in the airways.13 While DPPC can form a surface layer devoid of surface tension, it is not effective in vivo on its own; rather, it requires other lipids and especially surfactant-associated proteins to adhere to the lining of the lung. These proteins are also important in modulating inflammation and immune responses in the lung.14 The proteins discovered to date have been named surfactant protein (SP)-A, SP-B, SP-C, and SP-D; without SP-B, in particular, the individual cannot survive.15
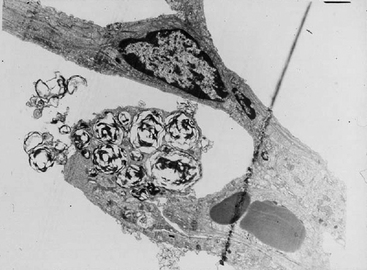
The lamellar bodies of type II pneumocytes make and store surfactant (Figure 8-6). The surfactant is subsequently released into the airways by the process of exocytosis, at which point, DPPC forms a lattice structure called tubular myelin in the presence of SP-A, SP-B, and Ca2+. DPPC is an amphipathic molecule, and its polar head associates with the aqueous phase lining the airways and acts as a tether for the molecule.16 The lipid portion faces the alveolar air; it is this portion of DPPC that resists film collapse at end expiration, and thus resists lung collapse. The low surface tension also ensures that the net flow of fluid is from the alveoli to the interstitium, thus counteracting pulmonary edema. As alveoli enlarge and stretch, each DPPC molecule is further from the next, thus resisting film collapse less. In this way, surfactant confers greater ability to resist collapse on smaller alveoli, which are in need of this neat trick.15
Surfactant also contains four other phospholipids: phosphatidylglycerol assists the surface activity of PC, but the function of the others is not known. However, in the majority of mammals, phosphatidylinnositol is the primary acidic phospholipid during the early stages of lung maturation; this changes to phosphatidylglycerol when the lung is fully mature.15 Unfortunately, the horse does not seem to adhere to these general principles. Paradis and coworkers investigated the amniotic fluid of premature and term foals, and found that neither DPC/sphingomyelin ratios nor the presence of phosphatidylglycerol were dependable in differentiating between preterm foals with respiratory distress syndrome (RDS), preterm foals without RDS, and term foals, unlike in the human.16,17
HOW DOES SURFACTANT DIFFER BETWEEN PREMATURE INDIVIDUALS AND TERM INDIVIDUALS?
First, the absolute amount of surfactant in term infants is almost 10 times that of premature infants, and the time from synthesis to secretion is slower in prematurity.15 The surfactant is less biophysically functional in the premature individual, and is more sensitive to inactivation by inhibitors, probably because the protein level is decreased. Lung injury itself further decreases the alveolar pool of surfactant, and the surfactant’s biophysical function becomes yet more diminished. This cascade of events is further compounded by dilution of surfactant with edema fluid.18
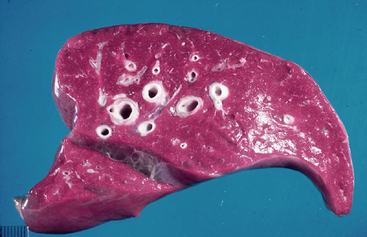
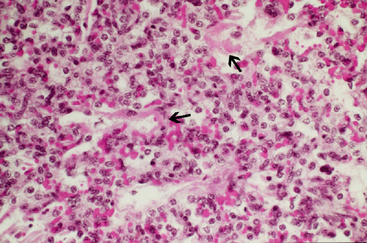
The most important thing to note is the effect of surfactant deficiency on respiratory mechanics in the preterm lung. When the clinician conducts pressure-volume curves on preterm lungs, they do not begin to open before 25 cm H2O. One of the biggest problems with surfactant-deficiency–associated lung injury is that there is massive heterogeneity in the lung. More collapsed units with smaller alveoli may require 40 to 50 cm H2O of pressure to open; this may generate sufficient volume to rupture other, more compliant units. The higher critical opening pressures and higher volumes in the few alveoli that remain inflated result in considerable damage, which is known as NRDS. Hyaline membrane disease refers to the sequela of NRDS found on pathology; these membranes form consequent to the exudation of protein and fluid into the alveoli, and grossly, the lungs appear liverlike (Figures 8-7 and 8-8).12 Surfactant allows us to avoid this sequela by decreasing surface tension, maintaining lung volumes, improving ventilation/perfusion matching, and thus oxygenation, all while decreasing the work of breathing.5
WHAT DO WE KNOW ABOUT SURFACTANT IN FOALS?
Soon after Pattle discovered the stabilizing properties of surfactant, he published an account of surfactant deficiency in a foal.19 Barnard and colleagues were successful in showing that the lungs of the foal show considerable maturation relatively early in gestation, and that alveolar development began by 260 days, or 80% of gestation, similar to humans, wherein alveolarization begins at approximately 84% of gestation.20,21 Type II pneumocytes, which produce surfactant, were clearly differentiated and contained osmophilic lamellar inclusions by 260 days, but nonsecretory forms were still seen at 320 days in some foals. By 320 days, alveoli were more numerous than the larger respiratory bronchiole spaces. When Barnard and colleagues looked at the elastin and collagen content of dysmature foals, they found that content of both was lower than in controls. Arvidson and coworkers demonstrated that an increase in total phospholipids occurs between days 100 and 150, and, finally, Pattle and coworkers demonstrated that surfactant begins to mature in the equine lung when approximately 88% of gestation has been completed; however, maturation is generally not complete until full term, and before full term, the activity of existing surfactant may be reduced.22,23
WHAT IS THE EVIDENCE FOR SURFACTANT DEFICIENCY IN PREMATURE FOALS?
In one study of naturally occurring premature foals, Rossdale found that the lungs of four of the foals showed evidence of severe atelectasis, macroscopically the lungs appeared red and liverlike in consistency, and that cut pieces sank in fixative.24 The bubble method, which has now been superseded by more sensitive and specific methods, demonstrated reduced surfactant activity in one foal.15 Macroscopic examination revealed widespread atelectasis, congestion in the capillaries, and desquamation of bronchiolar epithelium.24 Dubielzig observed pulmonary lesions of neonatal foals that died at less than five days of age, and found patchy atelectasis in 35% (some of which may have been due to NRDS) and hyaline membranes in 5.4%. In another study, hyaline membranes were observed on microscopic examination.25 Indirect evidence for surfactant deficiency in premature foals is also found in clinical studies of induced premature foals. When Rose and coworkers looked at foals induced between 270 and 320 days, the most striking clinical finding was respiratory distress and hypoxemia from the moment of birth.26
A second study by Rose and Hodgson examined the response of premature foals to oxygen, and found that they had a very poor response to nasal insufflation of oxygen, implying a high degree of shunt, likely intrapulmonary.27,28 After this promising start, an extensive review of the literature reveals that the study of surfactant in the term and premature foal has been largely neglected and, although we can infer from our knowledge in other species that respiratory distress and failure in a premature foal is most likely associated with surfactant deficiency, more extensive pathologic and physiologic studies must be conducted in order to fully to understand this problem in the foal.
The initial workup on ’03 Surf’s Up included arterial blood gas, chemistry profile, immunoglobulin assessment, and blood cultures. The CBC revealed neutrophilic leukocytosis (15,000 WBC/μl, 13,000 segmented neutrophils/μl) and elevated fibrinogen of 600 mg/dl. The chemistry profile revealed mild azotemia (creatinine 3.2 mg/dl). Her immunoglobulins were between 400 and 800 mg/dl, reflective of the colostrum administered by the referring veterinarian. The foal was normoglycemic. Her blood gases revealed marked hypoxemia (48 mm Hg) with mild hypocapnia (32 mg/dl) and a normal pH (7.34). Her calculated sepsis score was 14.
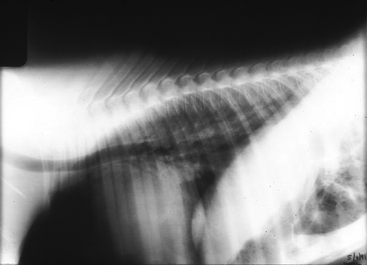
Radiographs of the chest revealed a mild diffuse interstitial pattern (Figure 8-9). Radiographs of the carpi and tarsi revealed poorly ossified cuboidal bones. There was no growth on blood cultures.
DIAGNOSTIC TESTS
The high neutrophil count and fibrinogen level indicate that ’03 Surf’s Up had been subjected to considerable inflammatory insult in utero. The high sepsis score supported a suspicion that the inflammation was accompanied by a possible active infection. The radiographs were suggestive of pneumonia and possible surfactant deficiency or functionally immature surfactant.29 Both before and after oxygen, the A-a gradient was consistent with considerable V/Q mismatch, as well as mild shunt, either intra-pulmonary secondary to partial atelectasis or a focus of pneumonia, or potentially due to pulmonary hypertension with reversion to fetal circulation. The FRC was mildly reduced, consistent with moderate restrictive lung disease, such as pneumonia or patchy atelectasis. The mildly elevated creatinine may have reflected placental pathology, prerenal disease, or intrinsic renal disease. The extent of prematurity was attested to by the poor cuboidal bone ossification. Nonetheless, ’03 Surf’s Up was not showing severe signs of respiratory distress. How can this be explained?
LUNG MATURITY AND INFLAMMATION
Chorioamnionitis, which correlates with pro-inflammatory cytokines IL-6, IL-1B, and TNF-alpha in amniotic fluid, is common in human preterm births, and is highly associated with decreased risk of RDS.21 Recent studies in rabbits show that pro-inflammatory cytokines can influence lung maturation.30 These investigators also found that IL-1 alpha increased surfactant lipids, and improved pressure-volume curves in a dose-dependent fashion. Moreover, in the lamb, the fetal lung responds to injection of endotoxin in utero into amniotic fluid, by increased mRNAs for SP-A, SP-B, SP-C, and SP-D within one day. In this study, SP mRNAs increased 100-fold over control values by seven days; DPPC increased in concert, linearly.31 Similarly, LeBlanc and colleagues found elevated levels of pro-inflammatory cytokines in the amniotic fluid of mares with ascending placentitis.32 The ability of the fetal lung to mature in response to inflammatory cytokines may explain the clinical impression of many internists that the endogenously premature foal has a better overall chance of survival than does the exogenously premature foal.
The initial workup on ’03 Last Gasp was similar, but the results were quite dissimilar. The CBC revealed neutropenic leukopenia with mild lymphocytosis, with 1500 neutrophils/ml and 3,000 lymphocytes/ml. Fibrinogen was 200 mg/dl. IgG was 400 mg/dl, indicating partial failure of passive transfer. Chemistry profile revealed marked hypercreatininemia (5.2 mg/dl) and hypoglycemia (40 mg/dl). An arterial blood gas revealed PaO2 of 37 mm Hg, PaCO2 of 50 mm Hg, and pH of 7.20. The calculated A-a gradient was 53 mm Hg. Immediate nasal insufflation of oxygen resulted in an O2 tension of 95 mm Hg and PaCO2 of 75 mm Hg, resulting in a calculated A-a gradient of 65. FRC-helium measurements were stunningly low at 700 ml. Radiographs showed a marked alveolar pattern with air bronchograms throughout (Figure 8-10). A sepsis score was elevated at 18. Radiographs of the carpi and tarsi showed moderate cuboidal bone ossification.
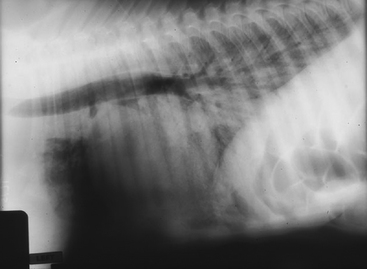
’03 Last Gasp had marked clinical evidence of respiratory distress and severe hypoxemia to match. Although his CO2 was only mildly elevated at presentation, in the face of tachypnea, it ought to have been somewhat low. The elevated PaCO2 after nasal insufflation of oxygen indicated that ’03 Last Gasp had lost his sensitivity to elevations in CO2 and was relying on hypoxic drive for the stimulus to breathe. When he received supplemental O2, he decompensated this hypoxic drive. The A-a gradient was markedly elevated, compatible with considerable V-Q mismatch. The venous admixture was compatible with shunt of intrapulmonary or cardiac origin, and in this case, we suspected that there might be secondary pulmonary hypertension present. The very low FRC measurements were concerning, as it reflected serious loss of end-expiratory lung volume. Although the ossification of the cuboidal bones was compatible with a foal that was not technically premature, the severe, diffuse, alveolar pattern was strongly suggestive of primary surfactant deficiency.29 The CBC was also compatible with prematurity; unlike ’03 Surf’s Up, this foal had neither neutrophilia nor elevated fibrinogen. Instead, the leukogram was suggestive of lack of adrenocortical influence in producing the more usual elevated neutrophil-to-lymphocyte ratio in the normal term foal. The neutropenia may also have been reflective of sepsis. The hypercreatininemia was compatible with intrinsic renal disease, placental pathology, or transplacental passage of elevated maternal creatinine.
Veterinarians are considerably hampered in their ability to monitor and predict prematurity in foals. Moreover, even if we have a strong clinical suspicion that the foal may be born premature, as with ’03 Surf’s Up, we lack good tests for determining whether the foal’s respiratory system will be mature. The most commonly used method for determining respiratory maturity in humans is the lecithin (DPPC) to sphingomyelin ratio, however, it has relatively poor specificity and sensitivity.16 Unfortunately, one of the better tests in humans, the lecithin to PG ratio, has shown conflicting results in horses.17 Until we have more extensive research into the composition and function of surfactant in the equine neonate, it is unlikely that we will be able to progress in the prediction of lung maturity in the premature foal.
HYPOXEMIA BEYOND SURFACTANT
During fetal life, blood flow through the pulmonary circulation is low—less than 10% of cardiac output. Blood is effectively shunted away from the pulmonary bed because the fetus’s hypoxic state results in constriction of the pulmonary vasculature. In the normal foal, pulmonary vascular resistance drops dramatically, and essentially all of the cardiac output flows with ease through the pulmonary bed. This situation can be altered by both primary pulmonary hypertension, (known as “black lung” PPHN, and relatively rare in human neonates) or by secondary pulmonary hypertension (known as “white lung” PPHN, and commonly seen as a complication of pulmonary vasoconstriction secondary to hypoxia in neonates).33 The monikers black lung PPHN and white lung PPHN refer to the presence of infiltrates seen on chest radiographs in secondary PPHN, versus the typically clear radiograph seen in primary PPHN. It is important to image the lung either with radiographs or computed tomography; however, without this critical information it is impossible to determine if the PPHN is of primary origin or, as is much more common in human neonates, secondary to parenchymal disease.34 Cardiac ultrasonography can be useful in further documenting whether there is right-to-left shunting of blood across the ductus arteriosus or foramen ovale, and to estimate right-sided vascular pressures.
Treatment for PPHN comprises primarily oxygen supplementation and frequently mechanical ventilation, in addition to treating acidosis, which may reinforce the abnormal circulatory pattern. Nitric oxide (NO), a vasodilator, has also been used successfully in infants with pulmonary hypertension, as well as in cases of suspected PPHN in foals.34,35 Sildenafil, a common drug used for erectile dysfunction in men, is a phosphodiesterase-5 inhibitor that has also been used in foals to treat with PPHN (personal communication Dr. Pamela Wilkins).



