Fig. 1
Steps in next-generation sequencing and data analysis workflows where error and bias introduction may occur
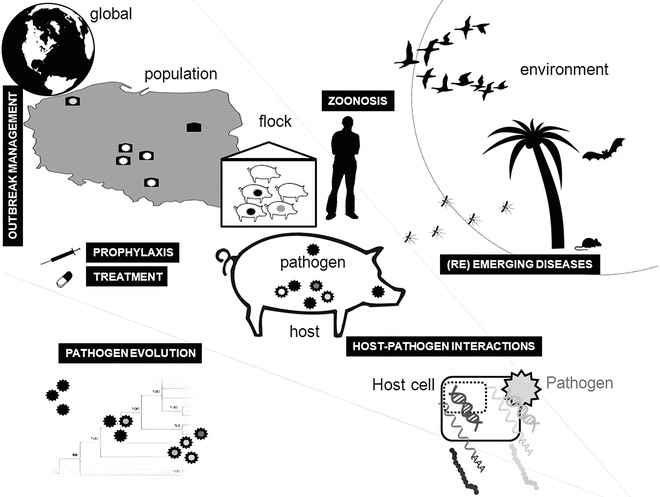
Fig. 2
High-throughput technologies can be applied to numerous aspects of animal infectious diseases
As a result of errors and bias introductions, NGS data needs to be “cleaned”. This includes sequence filtering (removing low-quality sequences) and alignment followed by variant calling and error correction. Discriminating true biological variants from those due to experimental noise is an important issue when trying to identify low-frequency variants in a population, for example, in viral quasispecies or metagenomic analyses, and there are currently a number of bioinformatics tools to aid in this (e.g., [23–26, 19]). Currently, a multitude of software has been developed to address different aspects of NGS analyses [27, 28]. However, the available algorithms for both genome assembly and amplicon analysis can present some limitations [29], meaning that custom-made scripting and in-house resolution of bioinformatic problems are often needed to investigate novel datasets and specific hypotheses. In this context, researchers are frequently faced with the need to acquire computer skills and bioinformatics expertise. To evaluate the potential of NGS for a wider group of scientists and diagnosticians, there is a real need to develop flexible and practical bioinformatics workflows that can provide user-friendly tools for the analysis of massive datasets and that become publicly available. Although some software with a menu-driven approach is available (e.g., Geneious, CLC Workbench, Galaxy), most applications are optimized on UNIX-based operating systems and require some bioinformatics expertise. Although less user-friendly, UNIX-based pipelines are typically freely available to the NGS user community and are equipped with algorithms that track the high pace of innovation in the NGS field.
A further issue is the scale of genetic data produced by NGS technologies which presents a physical constraint in terms of data storage and analysis. Although limited datasets (e.g., resulting from the desktop-range 2nd-generation sequencers) can be managed using modest computing resources, like high-end desktop computers running virtual Linux machines [4], larger datasets typically require high-performance computational clusters, which present a considerable investment and require sufficient information technology (IT) support. Cloud computational resources (i.e., renting time from commercial high-performance computational clusters) may be a solution [30] although further developments are needed, given the data transfer issues resulting from huge file sizes [4]. Another issue for diagnostic laboratories is protection of data from unauthorized access, which cannot be guaranteed in the cloud, as data from diagnostic examinations need to be kept confidential. For labs frequently producing NGS data, data storage and backup costs can be substantial. Ideally, these huge genetic datasets should be made publicly available to the scientific community as they provide a source of information applicable to better understanding disease, design of targeted assays, systems biology, and integrated OMICS analysis approaches. To this end, online repositories such as the Sequence Read Archive (SRA; [31]) have been created to store both raw NGS and intermediate analysis files.
It will also be important to consider how results from complex and massive NGS datasets will be communicated to policy groups and the public and become a decision-supporting tool. To this end, it is necessary that scientists and diagnosticians develop and agree on data formats for the communication of NGS results for analyses that go beyond simple genome sequences, for instance, for reporting quasispecies compositions.
3 Application of NGS to Animal Infectious Disease
NGS technology is now being increasingly applied to study the etiology, genomics, evolution, and epidemiology of animal infectious diseases as well as host-pathogen interactions (Fig. 2). These applications have provided novel insights and illustrate the potential of this new technology to directly impact on our understanding and control strategies for animal infectious disease.
NGS platforms have been instrumental in the completion of large animal genomes and the documentation of genomic variation (reviewed in [32]). Available livestock genomes now include bovine, pig, sheep, equine, and avian [32] which provide an important source of knowledge for understanding food production and animal interaction with infectious pathogens. Additional livestock genome sequencing efforts have documented genomic variation providing information for the development of genetic markers applicable to animal breeding genetics [33–35], including traits related to pathogen resistance and interaction with microbial communities in poultry [36]. Others have used novel sequencing technologies for the targeted study of specific gene families occupying key roles in host immunology (e.g., Toll-like receptor (TLR) gene family [37]).
The high variability and large size of the mitochondrial genome (mtDNA) of eukaryotic parasites have been recently explored using NGS (reviewed by [38]). mtDNA sequences proved very informative in epidemiological studies [38] but also include comparative mtDNA sequencing of parasites with low and high zoonotic potential [39]. Targeting specific polymorphic genes in the Cryptosporidium parvum genome using NGS, extensive intra-host genetic diversity was documented [40]. Studies of the transcriptome (all mRNA transcripts in an organism, tissue, or cell; also called RNA-Seq) of different parasite species and/or developmental stages provide insights into aspects of gene expression, regulation, and function, which are major steps to understanding their biology (reviewed in [41]). Examples include the characterization of the transcriptome from Eimeria sp. from chicken [42] and Taenia sp. from sheep [43]. In addition, RNA-Seq data have been used to predict potential drug targets [44] and to identify key genes involved in anthelmintic resistance [45].
Over the last five years, NGS has been used as an extremely important tool in the tracing of transmission, genome characterization, and outbreak management of both viral and bacterial diseases. The sequencing of these two pathogen types poses very different sets of challenges and issues, where the large data output expressed typically in the Mb (megabase) to Gb (gigabase) range [4] is particularly suited for the sequencing of larger bacterial genomes. The high plasticity of some microbial genomes, with large mobile elements, gene-coding plasmids, chromosomal genes, and regions of extensive genetic variability, can frequently complicate genome assembly [46]. While most viral genomes are significantly smaller than their bacterial counterparts, the viral replication biology (particularly that of RNA viruses) poses its own unique problems. These involve the inherent variability of many viral genomes due to replication machinery lacking efficient proofreading mechanisms. This, combined with a short generation time and high replication rate, results in a complex mix of differing genomes (a “swarm” of closely related viruses) within a single host that are often termed as “quasispecies,” reviewed in [47]. In addition, recombination and reassortment of segmented viral genomes frequently occur. NGS techniques offer an unprecedented “step-change” increase in the amount of sequence data that can be generated from both types of these samples.
Figure 3 (different scales of sequence analysis) highlights where genetic analyses can target different biological scales and whether these are within an individual host, or between hosts, resulting in either host variation or inter-herd diversity/outbreak transmission.
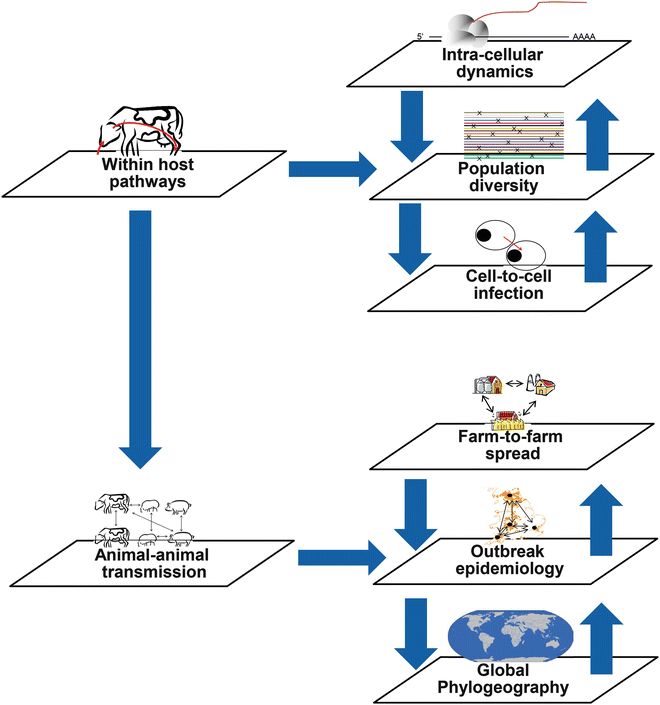
Fig. 3
The differing levels of intra- and inter-host variation that can be explored using NGS technologies range from intracellular dynamics to epidemiological applications
At the level of the quasispecies, NGS technologies can now determine complete viral genomes to a fine-point resolution, allowing the quantification of viral diversity within samples [48] and making the sequencing of large numbers of samples economically feasible. The technology will allow the comparison of genetically diverse populations from different replication sites within a host [49, 50]. Wright and colleagues investigated the genetic diversity and resulting quasispecies population after inoculation of foot-and-mouth disease virus (FMDV) into a single animal and identified genetically distinct populations originating from different lesions [50]. Morelli and colleagues [51] studied the evolution of FMDV intra-sample sequence diversity during serial transmission in bovine hosts, providing novel insights into the fine-scale evolution of an RNA virus. NGS can also provide insights on microevolutionary processes of viruses at different scales, including the fine-point resolution molecular epidemiology analysis of outbreaks [52].
Recent studies on influenza A viruses have demonstrated that minority variants present in the donor population can be successfully transmitted to the recipient host and become prevalent with unpredictable impact on the virus biological properties [53, 54]. These findings suggest that the use of NGS approaches in RNA virus surveillance will be strategic to promptly detect biologically relevant viral quasispecies and will help in expanding our understanding of viral dynamics and emergence and the possible implications of mutation emergence for studies done using isolated viruses [55, 56].
The study of the viral swarm within individual hosts also has implications for understanding the evolutionary dynamics of viral populations under selection pressures, e.g., antiviral drugs. This has been a particularly active field in human medicine, e.g., with regard to human immunodeficiency virus (HIV) antiviral drugs response, drug resistance, and viral tropism (reviewed in [57–59]) and human influenza A (e.g., [60]) studies. The technologies’ application to personalize antiviral treatment as a function of genetic marker makeup in human medicine is just around the corner [61]. Although at present only an emerging field in veterinary science, the development of antiviral drugs has the potential to translate into efficient animal infectious disease control strategies (e.g., [62, 63]).
The majority of the papers using NGS to investigate animal infectious disease focus specifically upon the level of animal-to-animal transmission and the characterization of pathogens within a single host, as this yields the most useful data in terms of outbreak management and identifying mechanisms/sources of disease transmission. For example, Lefébure and colleagues [64] used NGS to study genome complexity and horizontal gene transfer in foodborne Campylobacter spp. Biek et al. [65] studied local transmission patterns of Mycobacterium bovis in cattle and wildlife reservoirs using whole genome sequences from 31 samples originating on five farms. These demonstrated enough diversity between individual outbreaks to determine evolutionary variation down to herd level. The identification of novel antimicrobial resistance genes in the foodborne pathogen Campylobacter coli [66] was possible using NGS, and the application of NGS technologies during a recent crisis involving foodborne enterohemorrhagic Escherichia coli O104:H4 allowed a swift genomic identification [67] that was key to the management of this crisis. Finally, the technology also proved very informative to study the molecular epidemiology and evolutionary history of extremely monomorphic Mycoplasma mycoides subsp. mycoides SC [68] in addition to studies tracing medically significant pathogens [69–71]. Samples from the US 2006–2007 West Nile virus (WNV) outbreak in birds were characterized using Illumina sequencing, resulting in the identification of a new genetic variant containing a 13-nucleotide indel [72]. A survey of Chinese domestic fowl using RNA-Seq on an Ion Torrent identified a novel Coronavirus, providing insights into the diversity and distribution of avian coronaviruses [73]. A further study has also investigated the role of Usutu virus in causing epizootic infection in blackbirds in Germany [74].
The advent of NGS has also led to the cost-efficient sequencing of complete viral genomes including avian influenza virus [75–78], classical swine fever virus [79], and bluetongue viruses [80]. An optimized method incorporating 454 sequencing for universal nonspecific RNA viral genomes from brain and cell culture material was applied to Lyssaviruses [81]. Other groups have reported the characterization of Louping ill virus in lambs [82], porcine reproductive and respiratory syndrome virus [83], and herpesviruses from Asian elephants [84]. Furthermore, studies using random amplification techniques have identified mixed infections of paramyxoviruses and avian influenza in bird populations [77, 85, 78]. Efficient influenza A-specific resequencing strategies [86, 87] have allowed the study of quasispecies-scale genetic variability with implications for immune response [88, 89], host cell line adaptation [90, 91], antiviral drug resistance [92], and pathogenicity [53]. Likewise, efficient targeted CSFV genome sequencing using NGS has led to insights in classical swine fever virus (CSFV) epidemiology based on isolates from an outbreak in wild boar from Germany [79] and in the role of quasispecies diversity in CSFV pathogenicity [93].
NGS technology has also allowed the characterization of complete microbial communities without prior knowledge. For instance, the unbiased characterization of conserved bacterial ribosomal RNA-encoding sequences (rRNA profiling) has been applicable to whole microbial community characterization (e.g., [94, 95]) and to molecular characterization of (uncultured) bacteria [96]. Metagenomics is the determination of the sequence content of a complete microbial community (reviewed in [97]). The analysis of the resulting data can be taxonomy oriented (identification and quantification of species diversity; [98]) or function based (identification of coding gene diversity, e.g., [99]). The latter has significant potential, e.g., in the screening for virulence-associated, antibiotic resistance genes, and vitamin production-associated genes in microbial communities [100]. NGS also offers the potential of unbiased sequencing of the nucleic acid content of a sample and has been applied to the characterization of the viral metagenome in samples [101] or the identification of unknown or unexpected viruses in diseased animals or insect vectors. Furthermore, metagenomic NGS workflows allow the study of the interaction of treatment with an animal’s microbiome [102]. In the microbiology lab, NGS has the potential for greater diagnostic resolution than any other typing method, and clinical microbiology labs are currently investigating its potential for routine diagnosis [103, 104].
Using NGS-based metagenomic approaches, multiple potential disease agents have been identified in a wide range of both domestic and wild animals (reviewed in [105–109]). Although the common goal is to identify potential pathogens, the studies can roughly be divided into three categories: (1) investigations of outbreaks of unknown etiology, (2) investigations of well-known disorders presumed to be of multifactorial etiology, and (3) metagenomic studies of reservoir species and vectors. Examples of the first category include the identification of a novel Orthobunyavirus affecting cattle (described in more detail below), an astrovirus in the brain of farmed minks suffering from encephalomyelitis [110], and a novel picornavirus as candidate etiologic agent for turkey viral hepatitis [111], among others. The second category encompasses investigations aimed at finding contributing infectious agents to complex diseases, such as colony collapse disorder of honey bees [112, 113] and postweaning multisystemic wasting syndrome in pigs [114]. Studies in the third category have been performed on diverse animal species suspected to be important reservoirs, such as bats [115, 116], African bush pigs [117], and red fox [118], as well as typical vector organisms, such as ticks [119].
Although it is an important first step, the identification and genetic characterization of candidate pathogens are not enough to establish causal relationships or understand how they may be associated with disease. It is therefore necessary to use a synergistic approach combining molecular diagnostic tools, such as NGS-based metagenomics and follow-up PCR-based assays targeting detected pathogen sequences, with more conventional diagnostic methods, including isolation and characterization. This is crucially important in situations where metagenomic data indicate the potential presence of multiple pathogens. While PCR-based prevalence studies in matching disease cases and healthy controls can provide further evidence for disease association, isolation of candidate pathogens is required to assign causality by addressing Koch’s postulates [120]. The assembled data from such a multidisciplinary (pathology, epidemiology, metagenomic data, PCR prevalence studies, isolation, characterization, etc.) should be used to identify the most likely candidate etiologic agent and to make informed intervention decisions. The synergetic and parallel use of molecular and classical methods not only results in detection of infectious agents and development of targeted diagnostic tests but also has the potential to make isolates or strains available shortly after the occurrence of outbreaks. The availability of isolates or strains is of special importance to allow the design of effective vaccines or antimicrobial drugs.
The power of NGS to boost the veterinary laboratory community’s responsiveness to emerging diseases was demonstrated through the discovery of a novel Orthobunyavirus in 2011 associated with fever, decreased milk production, and diarrhea in dairy cattle. Metagenomics, using 454 technology, allowed the identification of a novel virus, subsequently named Schmallenberg virus (SBV), in an epidemiological cluster of diseased cattle in Germany [121]. These viral sequences were used to rapidly design targeted molecular tests that were used to confirm a clear association between the presence of the virus and affected animals [110]. International adoption of these molecular tests identified a widespread occurrence of SBV in European countries (http://www.efsa.europa.eu/en/supporting/pub/429e.htm) and its detection in stillborn and malformed lambs [122, 123], as well in insect vectors [124, 125]. The molecular tests were also helpful in targeting samples for isolation of the virus, which ultimately led to the development of a prototype vaccine currently under evaluation [126].
Metagenomic NGS workflows also have the potential use for quality control of biological products [127] and vaccines [128–132] and provide a powerful approach for the identification and characterization of unexpected of highly divergent pathogen variants [133, 85] that may remain undetected using targeted diagnostic tests.
The technological possibility to study both the host and the pathogen with high resolution on the level of their genome, transcriptome, or proteome opens opportunities to study host/pathogen interactions at several levels ((genomics, transcriptomics, microRNAs (miRNA)) and ultimately to analytically integrate these levels (integrative omics or systems biology) aiming to study the interaction of pathogen, microbiome, and host biological networks with many examples in veterinary science. Nordentoft and colleagues [134] used NGS metagenomics to study the influence of livestock management parameters and infection with Salmonella enteritidis on the microbial community in the chicken intestinal tract. Another study [135] documented the effect of Campylobacter jejuni infection on the chicken fecal microbiome. The application of metagenomic techniques in poultry production could lead to the development of novel alternatives to antibiotic growth promoters and better understanding of the colonization of food production animals by foodborne pathogens such as Salmonella enterica and Campylobacter spp. [36]. Other studies investigated the host response to pathogen infection. Glass and colleagues [136] used NGS transcriptomics to document bovine resistance and tolerance traits to parasitic infection. The technology was also used to study the ferret transcriptome response to influenza infection [137], the chicken transcriptome response to Marek’s disease [138], the swine response to porcine reproductive and respiratory syndrome virus infection [139], and the changes in the mouse transcriptome after Brucella sp. infection [140].
microRNAs are considered to be a key mechanism of gene regulation in both parasites and viruses. Their characterization contributes to better understanding the complex biology of pathogens. Wang and coworkers [141] characterized microRNA sequences from Orientobilharzia turkestanicum, a fluke with zoonotic potential infecting sheep, and identified key target miRNAs for parasite energy metabolism, transcription initiation factors, signal transduction, and growth factor receptors. Virus-encoded microRNAs (vmiRNA) regulating viral or cellular transcripts can be targeted for virus discovery [142, 143]. miRNAs also play important roles in regulating host-pathogen interactions. NGS has been applied to investigate whether infection can modulate miRNA biogenesis and has also been used to identify miRNAs that influence pathogen replication, tropism, and pathogenic potential [144–149]. In particular, cellular miRNAs have been shown to interact with the viral genomic RNA or mRNA, facilitating or inhibiting the virus life cycle. These molecules have demonstrated immense potential as a source of antiviral therapeutics effective against a number of viruses (adenovirus, rabies, Venezuelan equine encephalitis, porcine reproductive and syndrome virus [150–153]) or for the design of live-attenuated virus vaccine based on miRNA-mediated gene silencing [154, 155, 147].
4 Conclusions
Next-generation sequencing technologies have the potential to revolutionize our understanding of the complex dimensions of animal infectious disease and infection biology (Fig. 2), ranging from the intracellular interactions to disease epidemiology. The application of high-throughput biotechnology platforms in these fields and their typical low-cost per information content has increased the resolution with which these processes can now be studied.
We now have high-resolution tools that provide veterinary diagnostic laboratories with the ability to undertake swift and flexible responses to emerging infectious diseases and unexpected pathogen variants. Moreover, these tools provide an increased resolution for the characterization of pathogens and provide important assets to improve our understanding. Fundamental research on pathogen evolution, adaptation, and virulence determinants can now be studied on a scale allowing within and between host dissections of genetic variability. Moreover, high-throughput tools open new perspectives to study the complex interaction between pathogen, host, and microbiome with very high resolution and to deepen our understanding of the key biological processes leading to protective immunity.
Not only will our increased understanding of pathogens and their interaction with livestock impact on future disease prevention, control, and management strategies, but the technologies may themselves become part of the intervention strategies, providing high-resolution data for molecular epidemiology to rapidly trace the origin and spread of outbreaks, for molecular typing, for predicting, and for optimizing the outcome of targeted treatment with antibiotics, antivirals, and anthelmintic.
The ready availability of high-resolution genomic and transcriptomic data will impact upon the targeted development of novel vaccines and drugs [156, 157], while NGS has the potential to become a powerful tool for the control of vaccines and other biological products.
As with any new technology, challenges remain. In the case of NGS, these include the requirement for expertise in both the laboratory and in the analysis of huge datasets and the current need for high investment in laboratory and data analysis hardware. As the technology is ever evolving towards lower cost, user-friendliness, and accessibility for smaller research and diagnostic labs, efforts are needed to make the data analysis more accessible to nonexpert users. This includes proper modeling of the sources of error introduction, solutions for public data storage, development of user-friendly but high standard analysis pipelines for routine applications, etc. Both the industry and the NGS user community can play a role in this evolution.
Similarly, recent improvements in protein and peptide separation efficiencies and highly accurate mass spectrometry have promoted the identification and quantification of proteins in a given sample [158]. Directly targeting peptide and protein content in a sample, proteomic approaches provide important additional information taking known issues, such as the quantitative discrepancy between mRNA transcript levels and final protein levels and posttranslational modification, into account [159].
Novel proteomic approaches have been applied to animal infectious disease research, including the study of E. coli response to chicken sera [160], proteomic profiling of porcine sera after FMDV infection [161], host-pathogen interaction during bovine mastitis [159], and metaproteomic studies characterizing the collective proteome of microbial communities [162].
This section contains excellent contributions exploring the application of high-throughput technologies to animal infectious diseases, including functional genomics of tick vectors infected with eukaryotic parasites, metagenomic approaches to detect bee viral pathogens, proteomics of vector-host-pathogen interactions, and NGS applications exploring parasites and intervention strategies.
Acknowledgments
The collaboration between the authors was supported by Epi-SEQ: a research project supported under the 2nd joint call for transnational research projects by EMIDA ERA-NET (FP7 project nr 219235). Additional support for this work in the United Kingdom was obtained from the Department of Environment, Food and Rural Affairs (Defra project SE2940) and BBSRC (BB/I014314/1).
References
1.
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467PubMedCentralPubMed
2.
Mullis K, Faloona F, Scharf S et al (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51:263–273PubMed
3.
Bartlett JM, Stirling D (2003) A short history of the polymerase chain reaction. Methods Mol Biol 226:3–6PubMed
5.
Radford AD, Chapman D, Dixon L et al (2012) Application of next-generation sequencing technologies in virology. J Gen Virol 93:1853–1868PubMedCentralPubMed
6.
Pareek CS, Smoczynski R, Tretyn A (2011) Sequencing technologies and genome sequencing. J Appl Genet 52:413–435PubMedCentralPubMed
7.
Loman NJ, Misra RV, Dallman TJ et al (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30:434–439PubMed
8.
Schadt EE, Turner S, Kasarskis A (2010) A window into third-generation sequencing. Hum Mol Genet 19(R2):R227–240PubMed
9.
Eisenstein M (2012) Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotechnol 30:295–296PubMed
10.
Brodin J, Mild M, Hedskog C et al (2013) PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS One 8:e70388PubMedCentralPubMed
11.
Kim KH, Bae JW (2011) Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl Environ Microbiol 77:7663–7668PubMedCentralPubMed
12.
Rosseel T, Van Borm S, Vandenbussche F et al (2013) The origin of biased sequence depth in sequence-independent nucleic Acid amplification and optimization for efficient massive parallel sequencing. PLoS One 8:e76144PubMedCentralPubMed
13.
Pettengill JB, McAvoy E, White JR et al (2012) Using metagenomic analyses to estimate the consequences of enrichment bias for pathogen detection. BMC Res Notes 5:378PubMedCentralPubMed
14.
Lysholm F, Wetterbom A, Lindau C et al (2012) Characterization of the viral microbiome in patients with severe lower respiratory tract infections, using metagenomic sequencing. PLoS One 7:e30875PubMedCentralPubMed
15.
Naccache SN, Greninger AL, Lee D et al (2013) The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J Virol 87:11966–11977PubMedCentralPubMed
16.
Whiteford N, Skelly T, Curtis C et al (2009) Swift: primary data analysis for the Illumina Solexa sequencing platform. Bioinformatics 25:2194–2199PubMedCentralPubMed
17.
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11:31–46PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


